Move those side-chains
Most developers of Virtual Screening software have succumbed to the temptation to ignore protein side-chain flexibility. Inevitably this means that hits are missed unless the contributions from VdW repulsions are reduced or hydrogens ignored. Unfortunately, one would expect these approximations to increase the numbers of false positive hits.
The increased computational power from Linux clusters and distributed computing has been used in recent years to increase the quantity of molecules screened. Little progress has been made in removing crude approximations or improving scoring functions. Docking algorithms which refine side-chain positions implicitly require a minimisation algorithm.
In our recent experience, Simplex is a poor choice of minimisation algorithm when refining side-chains. The reasons for this are two fold:
- The Simplex algorithm performs badly when rotating many side-chain bonds
- Each iteration requires the time-consuming intra-protein non-bonded energy to be recomputed.
THINK 1.32 includes a steepest descents minimiser to refine the ligand position, orientation, torsions and side-chain torsions. The following table shows some improved results over THINK 1.31 with NCI cancer cell line activity data:
| Protein | True positives | False positives |
| 1o4p-q1 | 13/14 | 38/68 |
| 1nvs-q1 | 10/13 | 32/61 |
These preliminary results used our revised scoring function with an additional penalty term for gaps between the ligand and the protein. In this example, extended protein atom radii were used in preference to explicit hydrogens which would be better but slower!
Find-a-Drug update
After nearly 300 queries searched, each with up to 0.5 billion drug-like molecules, and over 10,000 CPU years we have stopped generating data. We continue to pursue existing and new collaborations.
Screening without proteins
For a set of active molecules, the pharmacophores which occur most frequently in a pharmacophore profile can be used as search queries to generate the 3?D coordinates for conformers fitted to that pharmacophore. If each molecule has 2 conformers which exhibit the pharmacophore then for 8 active molecules there are 256 permutations of conformers, each of which has a corresponding union volume map. In general, it is reasonable to assume that for each pharmacophore the most credible union volume map is the one with the smallest volume corresponding to different molecules occupying the same space.
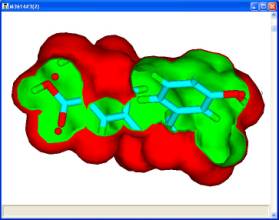
The union volume map can be used as a constraint in a pharmacophore search of a database or library. The use of such constraints eliminates a significant number of false positives during a search. Such maps which represent the volume used by active molecules are known as active volume constraint maps or ligand maps.
Towards a better scoring function
THINK uses an enhanced ChemScore function with
- An intermolecular VdW term
- A lipophilic-hydrophilic solvation term that penalises bad contacts
- A charge dipole model to supersede the empirical hydrogen bond term
- An improved ligand torsional entropy term.
In our experiments protein side-chain torsional entropy offered little improvement.