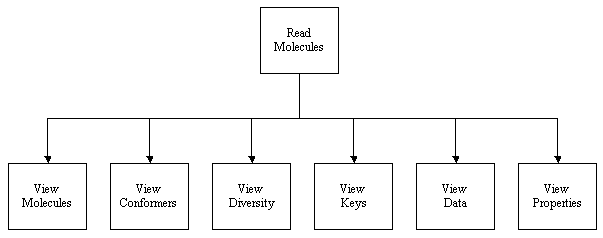
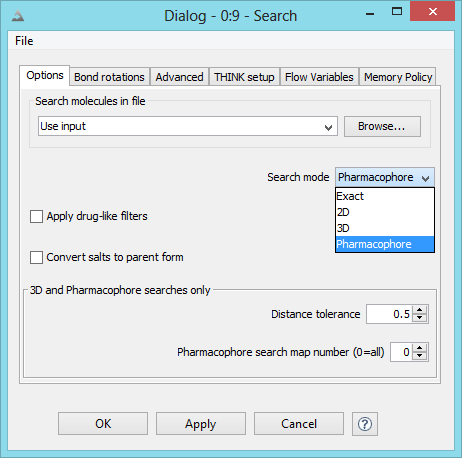
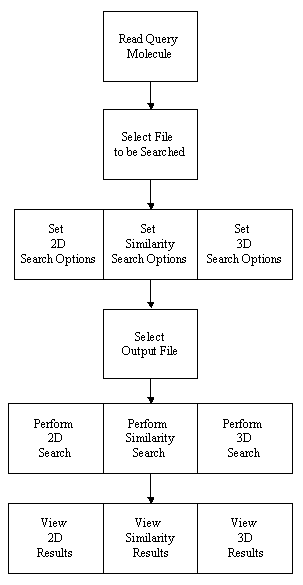
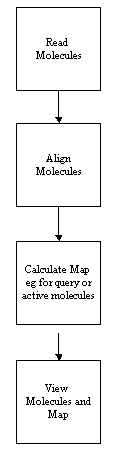
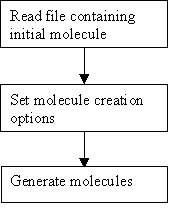
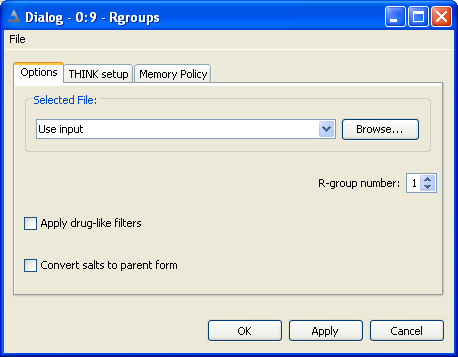
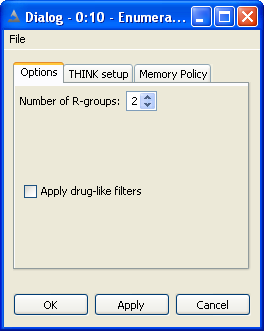
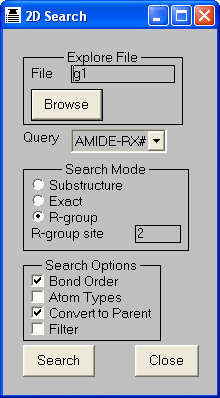
THINK is a modular system designed to assist with drug discovery lead generation
and lead optimisation by:
Structure-based virtual screening
This is a subset of Virtual Screening which is sometimes referred to as Docking, which finds molecules which can interact
with protein receptor sites. The software also has 2D, similarity and 3D searching functionality.
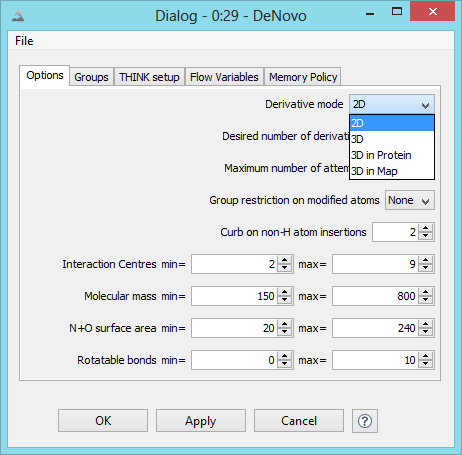
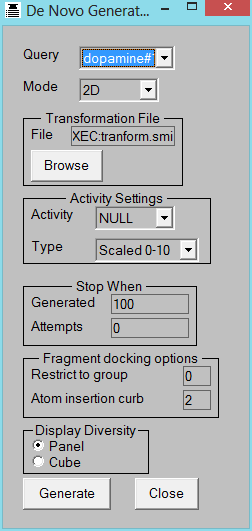
De Novo Derivative Generation
This operates (a) in 2D generating drug-like derivatives of a starting molecule or (b) in 3D when the core of the starting
molecule is retained and different substituents are created to fill either a protein site or a volume constraint map.
Pharmacophore profiling
Generating the list of pharmacophores which can be exhibiting by individual molecules or a pharmacophore profile for a set of molecules.
These can be used to select focused or diverse selections of molecules.
Data analysis
There are a range of standard KNIME nodes for data analysis.
Reviewing data, such as HTS results, to gain knowledge about desirable
and undesirable features of molecules.
The software can be used:
In text mode when commands are entered at the keyboard or read from executing scripts. This provides
full access to all the functionality of the software.
Interactively under Microsoft Windows using a series of Dialogs which are designed to present the most commonly used options within each facility.
Inconjunction with the KNIME workflow software for which a
series of plugin nodes are provided. KNIME workflows are normally used to
process a series of molecules. This provides most of the flexibility and functionality of command scripts with the benefit of
visualising the process.
The recommended mode of usage for new users is KNIME. The reader may switch the mode of this User Guide
to read information about the program's usage and examples relevant to each mode using the mode select controls.
Those sections of this manual which are mode specific have a letter prefix: K for KNIME; D for Dialogs and T for Text commands.
The THINK system currently consists of the following modules:
Module
Provides
Core module
Import and export of molecules via files; listings of molecular data; command
scripts
Graphics/GUI module (currently only available for Windows)
Displays 2D and 3D representations of molecules; diversity plots; functional
group distribution plots and histograms. The GUI provides a series of dialogs
that may be used in place of commands
2D module
Genetic algorithm to generate de novo derivatives of a starting molecule; tertiary
SAR analysis to create rejection criteria for the genetic algorithm; property
diversity calculation and selection; 2D substructure and similarity searching
3D module
Conformer generation; 3D coordinate generation (including fused rings); 3D
searching
Pharmacophores
Calculation pharmacophore lists and profiles; site searching (in conjunction with
3D module)
Usage Mode:
1.1 User Interface
THINK can be started from the Windows menu START > All Programs > THINK or by double clicking on
think.exe in the Windows Explorer. When THINK starts the console and explorer windows open.
THINK uses a console window to enter commands or open dialogs. This is the
main window used by the program. Additional windows will be created when necessary
to display molecules, data or perform searches.
Items on the dialogs may appear as:
Push buttons
Radio buttons - sets of circular buttons denoting mutually exclusive options
(eg display style)
Check boxes - square boxes for items that may be independently toggled on
or off. The box is checked when the when toggled on
Choosers - drop-down sub-menus indicated by an arrow to the right of some text
enclosed in a box
Keyboard entry items
Lists - either individual or multiple selections are supported depending on the context.
Double-picking a name within the list is equivalent to a single selection and pushing the associated button.
Bubble help is available for dialog items, tools etc.
The THINK software can be used under Windows and Linux with the KNIME workflow environment. The THINK plugins are downloaded from
http://www.treweren.com and extracted into the plugins folder under the KNIME directory tree. It is also necessary to
create the environment variables THINK_EXEC as the path to the THINK software and THINK_WORKING as a path to a folder in
which temporary files can be created.
Molecular modelling or informatics procedures are created, configured and executed by users as a visual workflow in KNIME.
THINK using KNIME allows integration with in-house developed and other commercial tools such as those available
from TRIPOS and Schroedinger. The main capabilities are illustrated by the following videos (with sound commentaries):
As the THINK nodes are designed for use with native KNIME nodes and those provided by other third party developers,
applications might include using other nodes to create a table of molecules (which have connection tables in a
SMILES or SDF column) and the outputs might be connected to other nodes for statistical analysis or
further computation. KNIME allows much higher productivity than writing command mode scripts and using a
workflow enables visualisation of the computational steps in the modelling procedure.
Each facility or utility in THINK has its own command, which may take one or
more keywords to supply additional options. A keyword may take a value (eg to
supply a filename). Commands are case independent and may be specified in upper, lower or mixed case.
where items in [] brackets may be optional. A few commands (eg EXIT) do not
take any keywords. Spaces are required to separate the command from any subsequent
keywords, and between keywords. Any values required by a keyword must be specified
using an "=" between the keyword and the value, with no spaces, ie keyword=value.
If the keyword may take a list of values, these should be separated by commas,
again with no spaces, ie keyword=value1,value2.
OPEN FILE=capsaicin.smi
SUGGEST ... ACTIVITY=EC50 OPTIONS=HIGH,LOG ...
It is also possible to omit the keyword= for manditory keywords provided they are
specified in the same order as the help file. Advanced users may find this practice saves
typing but it should not be used in script files.
The HELP command will list all the commands. A full list of keywords and values
available for any command may be obtained by issuing the command HELP followed
by the name of the command for which information is required or a the command followed
by a question mark.
HELP
HELP SUGGEST
SAVE ?
THINK allows multiple commands to be specified on a single command
line separated by a semi-colon (;).
1.2 Error Messages
Errors encountered while running KNIME workflows can be obscure and nodes developed by different
third parties are likely to have different style error reporting.
If THINK detects an error, this is normally reported to the console window with an error number and
some explanatory text. This information is also written to the log file (see section 1.3).
When THINK detects an error it will display a pop-up error message containing
the error number, some explanatory text and three buttons: Continue, Cancel and
More. Picking Cancel will cancel the operation immediately; picking Continue will
allow the calculation to continue if possible. Picking the More button will display
a second pop-up error window giving more information about the error. This information
may not be very helpful to the user, but can be useful to the THINK developers
when determining the cause of the error. The second pop-up window contains Continue
and Cancel buttons; these have the same effect as their equivalents on the first
window.
All messages from the first pop-up error window are echoed to the THINK console
window and written to the log file (see section 1.3).
When THINK detects an error it will write the error message containing
the error number and some explanatory text.
All messages from the first pop-up error window are echoed to the THINK console
window and written to the log file (see section 1.3).
1.3 Log Files
All commands issued during the execution of a THINK node are written to a file named output.log creating
in the working directory except for the Display node for which the log file is named output2.log.
The log file is overwritten when the next node is executed.
All commands issued during a THINK session are recorded in a log file. This
is named "think[n].log" where n is a counter and is created in the current working directory.
The log file contains the commands
issued by the dialogs. A log file may sometimes be subsequently replayed as a script by issuing the
command "CALL xxx" where xxx is the name of the log file. However, in many circumstances
it will need to be edited first using a text editor.
Note that THINK will automatically create a new log file incrementing n in the name
"think[n].log" each time the program is started.
All commands issued during a THINK session are recorded in a log file. This
is named "think[n].log" where n is a counter and is created in the current working directory.
A log file may sometimes be subsequently replayed as a script by issuing the
command "CALL xxx" where xxx is the name of the log file. However, in many circumstances
it will need to be edited first using a text editor.
Note that THINK will automatically create a new log file incrementing n in the name
"think[n].log" each time the program is started.
1.4 Identifying Molecules and Atoms
Conventionally molecules are assigned names or identifiers and these are usually the unique column
identifiers in KNIME data tables. In addition, within molecules atoms have atom types, serial numbers
and sometimes different group numbers. It is occasionally necessary to understand the
molecule and atom specifications used by THINK.
Some dialogs require the user to identify a molecule. Occasionally a dialog requires
atoms to be identified by their atom specifications (eg when using
the ROTATE dialog rather than the mouse for rotation). To do this successfully as well as understand
some output from THINK
requires some understanding of the molecule and atom specifications used by THINK.
Many commands require the user to identify a molecule. Occasionally the user
is required to identify atoms by typing their atom specifications (eg when using
the ROTATE command). To do this successfully requires some understanding of the
molecule and atom specifications used by THINK.
Each molecule has a molecule name. This is either:
read from the SMILES or SD file along with the molecule
composed of the filename, a "#" character and a counter, eg CAPSAICIN#1.
The counter shows the position of that molecule within the SMILES or SD file
If the molecule is one of a set of conformers, its name will include a conformer
number enclosed within parentheses (), eg ASP(3). The molecule name may be extended
by the addition of an "@" character and the name of the file from which
the molecule was read, eg "CAPSAICIN#1@capsaicin". This may be used
to distinguish molecules with the same name that came from different sources.
The full specification for any atom consists of three parts: the atom, residue
and the molecule identifiers. If there is only one small molecule present, or
if the atom identifier can uniquely specify the atom, then the residue and molecule
identifiers may be omitted. The atom identifier has the form:
type(serial.group)name eg C(13) or H14
where type is the atom type or element symbol for the atom; serial
is the serial number, group is the group number and name is the
atom name. Parts of the identifier may be omitted if this does not cause ambiguity.
The shortest common atom identifiers are (serial) or name subsets.
The type and group fields are omitted unless they are required to uniquely identify
the atom. If the type field is supplied, it must be followed by the parentheses,
either with or without the serial number, to distinguish it from the name
field. If the group field is not supplied, the leading "." should
also be omitted.
If the molecule has been read from a PDB file the residue name, sequence number
and insertion code, and chain id will be stored; for molecules read from other
file formats these portions of the atom specification are set to blank strings.
This information is preceded by an underscore "_" and has the form:
residue(sequence)chain eg TYR(42)
where residue is the residue name, sequence is the sequence number,
including the insertion code if present (eg 370, 85A), and chain is the
chain id. Like the atom identify, portions of the residue identifier, eg the chain identifier,
may be omitted if they are not needed to specify the residue(s) required.
If the molecule identifier is also required to pinpoint an atom it should be
separated from the atom and residue identifiers by a "^" character.
Thus, the full atom specification is:
There is no distinction between atom and molecule identifiers in upper, lower or mixed case.
Note that THINK will report all filenames in lowercase when listed as part
of the molecule identifier, even if they were entered as upper or mixed case names.
Symbols (see section 1.5.1) may be used
to identify atoms or residues. This is a convenient method of identifying a group
of atoms in a single operation (eg the active site of a protein). The symbol must
be an array comprising one or more array elements, and each element contains a
separate atom or residue specification. Each array element is taken in turn when
the symbol is processed. The symbol name must contain more than four characters to
avoid confusion with atom or residue names (eg SITEA would be interpreted as a symbol
but SITE would be treated as a residue name).
The array is terminated by a blank or undefined element. When a symbol is
used for the first time, this will occur automatically. However, if an array symbol
is reused, and the new array contains fewer elements, it is important that a blank
element is specified after the last valid element to ensure that THINK will determine
the array length correctly. For instance, if the new array only contains three
elements, then the fourth element should be set to a blank value.
If the symbol is to be used in place of an atom specification (eg when defining
the bond for bond rotation) the array element may contain any portions of the
full atom specification. For instance, CA_TYR(33) would refer to the alpha carbon
in residue TYR(33); and (23:29) would refer to all atoms with serial numbers in
the range 23-29. However, if the symbol is to be used in place of a residue specification,
the array element must contain only the residue components of the full atom specification,
ie residue(sequence)chain. The leading underscore "_" character
must be supplied before the symbol name.
In the examples below the symbol names have been highlighted to distinguish
them from the rest of the commands. See section 1.5.1
for full details on creating symbols.
LET ATOM1(1) = CA_TYR(33)
LET ATOM1(2) = " "
LET ATOM2(1) = N_TYR(33)
LET ATOM2(2) = " "
ROTATE ABOUT=ATOM1-ATOM2 ANGLE=30
LET SITEA(1) = (19:24)
LET SITEA(2) = (45:47)
LET SITEA(3) = (63)
LET SITEA(4) = " "
MODIFY INTERACTIONS=_SITEA
1.5 Command Scripts and Symbols
The THINK command set includes simple control commands such as LET, WHILE and
IF to enable the user to create command scripts. Apart from LET commands, these
must be saved in files (they cannot be entered directly into the console window)
and are played by issuing the command "CALL xxx" where xxx
is the name of the script file. The "@" character may be used in place of CALL.
Commands within scripts may be in upper, lower
or mixed case. LET commands may be issued from command scripts or typed directly
into the console window.
Command scripts may be nested up to 10 levels deep. A nested script is invoked
by the command "CALL xxx", where xxx is the name of the
script. A nested script ends, and control is returned to the calling script, when
a RETURN command is encountered. All nested command scripts should finish with
a RETURN command.
If a command script is interrupted by typing <CTRL-C> or picking the
Cancel button, all script files are closed and control is returned to the THINK
console window.
1.5.1 Symbols and Operators
Command scripts support local and global symbols. Local symbols only exist
within the command script being executed (and any nested scripts below the current
level) and are deleted at the end of the script. Global symbols persist after
the script has terminated. Both types of symbol are set via the LET command: local
symbols use the form "LET a = b" whereas global symbols
use the form "LET a := b", replacing the
"=" with ":=". Note that spaces are optional around operators and the "="
or ":=". Text strings should be enclosed within double quotes "";
names of symbols may be enclosed within single quotes '' or left unquoted. However,
use of single quotes around symbols and spaces around operators is recommended to avoid ambiguities. Symbol names
may use any alphanumeric characters (A-Z, 0-9) but must not conflict with the
THINK commands (issuing the command HELP will generate a full list of commands).
The symbols P1 to P9 have a special meaning within command scripts (see section
1.5.2) and should be avoided for user-defined symbols.
A symbol (local or global) may contain a single scalar value or a one-dimensional
array. Each member of an array is known as an array element and is identified
by its position within the array (eg ATOMS(3) would be the third element in the
array ATOMS). Each element within an array is set by a separate LET command (unlike
some other scripting languages, there is no way to set the contents of the whole
array through a single command).
THINK can extract (but not set) substrings of any symbol or array element for
use in another operation. A substring is specified as the name of the symbol or
array element, followed by the range of characters required. If the first value
in the range is omitted or replaced with a "*", THINK will start at
the beginning of the string; if the second value is omitted or replaced with "*",
THINK will finish at the end of the string. For instance if the symbol ALPHA contained
the alphabet in a single text string, ALPHA(5:8) would return the string "EFGH";
ALPHA(:3) would return "ABC" and ALPHA(23:*) would return "WXYZ".
LET FILE = "capsaicin.smi"
LET X-ANGLE := 45.0
LET ATOMS(1) = (9)
LET ATOMS(2) = (15)
LET ATOMS(3) = (21)
LET SUBST = TEXTARRAY(3)(5:17)
The following arithmetic, string and bit operators are supported in
LET statements:
%
Modulus
^
Exponentiation
*
Multiplication
/
Division
+
Addition
-
Subtraction
.
String concatenation
? xx ~ yy
String substitution - replace xx with yy
&
Bitwise AND
!
Bitwise NOT
|
Bitwise OR
:
Bitwise EOR
The arithmetic operators are processed in the order they are listed above,
with exponentiation having the highest priority. The string substitution operator
is very rarely used in user-written command scripts, but is used extensively in
the scripts that generate the THINK dialogs. THINK also supports relational
operators - these are described in section 1.5.3. The LET command
may also be omitted where this does not cause any ambiguity.
LET J1 = J1 + 2
LET FACTOR := 'RANGE' / 'SIZE'
LET NEWFILE = 'NAME' . ".smi"
1.5.2 Script Arguments
Up to nine values may be passed as arguments to a command script. This allows
a single script to be re-used (eg on different molecules) without having to change
the script file before each repetition. The arguments are specified after the
name of the command script in the CALL command, and are separated by spaces. Any
text strings that include spaces must be enclosed in double quotes ""
to ensure the whole string is treated as a single argument.
Within the command script, the arguments are identified by the special local
symbols P1 to P9. Unlike other local symbols, P1-P9 only exist within the current
command script and are not inherited by any nested command scripts below the current
level.
CALL COUNT.LOG ASP ASP.SMI
LET TEXT = "Counting atoms in " . P1
OPEN FILE='P2'
1.5.3 Control Commands and Relational Operators
Normally the statements in a command script are executed in the order in which
they appear in the file. This order may be changed through the use of:
GOTO commands to jump to a label somewhere else in the file
IF-[ELSEIF-][ELSE-]ENDIF commands to execute statements if a condition is
true (the portions in brackets [] may be omitted)
WHILE-END commands repeatedly perform a set of statements while a condition
is true
The label used by a "GOTO xxx" command is identified by the
statement "LABEL xxx". The IF-ENDIF commands have the form:
IF condition_1 block_1
ELSEIF condition_2 block_2
ELSE block_3
ENDIF
where the statements in block_1 are executed if the condition condition_1
is true, otherwise the statements in block_2 are executed if condition_2
is true. If neither condition is true, the statements in block_3 will be
executed. Either or both of the ELSEIF-block_2 and ELSE-block_3
sections may be omitted; only the IF-block_1 and ENDIF sections are mandatory.
The WHILE-END commands have the form:
WHILE condition block_a
END
where the statements in block_a are repeatedly executed while condition
is true. Note that block_a must include a statement that makes condition
false, otherwise the loop will never terminate and THINK will not respond to any
further commands.
The conditions used by the IF-ENDIF and WHILE-END commands compare the values
of two symbols or constants using one of the following relational operators:
=
Equal to
!=
Not equal to
>
Greater than
>=
Greater than or equal to
<
Less than
<=
Less than or equal to
&
Logical AND
!
Logical NOT
|
Logical OR
:
Logical EOR
When the logical operations give a non zero result the condition is considered to be TRUE.
Care must be taken when comparing text strings (they are case-sensitive) and
when comparing real numbers using an "equal to" test. It is recommended
that real numbers are compared using the "greater than" or "less
than" relational operators.
1.5.4 Error Handling
If an error is encountered during a command script, THINK will execute an implicit
GOTO command and jump to an error location. This may be defined in one of three
ways:
a label called ON_ERROR, ie "LABEL ON_ERROR"
a label in the current command script whose name is stored in the symbol ON_ERROR.
For instance "LET ON_ERROR = ERROR1" would be used with "LABEL
ERROR1"
storing the value "CONTINUE" in the ON_ERROR symbol, ie "LET
ON_ERROR = CONTINUE"
Use of the ON_ERROR symbol allows the destination of the error jump to be changed
whilst the script is executing simply by changing the value of the symbol. The
special value "CONTINUE" indicates that the error should be ignored
and the THINK should execute the next line in the command script.
LIST INFO=MOLECULES
...
LABEL ON_ERROR
WRITE CONSOLE "Error listing molecules"
LET ON_ERROR = NOMOLS
LIST INFO=MOLECULES
...
LABEL NOMOLS
WRITE CONSOLE "No molecules present"
For more advanced error control it is possible to have a different labels for different errors. Each error message written
by THINK has a unique number and this can be used in an array of symbols such as setting ON_ERROR(101) to catch no molecules present
(error message number 101).
1.5.5 Input and Output
Text strings can be written to an external file, the THINK log file or the
console window through the WRITE command: "WRITE file text" where
file is the name of the file to receive the data and text is one
of the following:
a single text string with no spaces
a text string (which may contain spaces) enclosed in double quotes ""
the name of a symbol containing the data to be written
File may be specified as LOGFILE or CONSOLE to write data to the THINK
log file or console window respectively. If an error occurs whilst the data is
being written out, THINK will execute an implicit GOTO command and will jump to
the FILE_ERROR location. This is analogous to the ON_ERROR error location (see
section 1.5.4) and may be an explicit label ("LABEL
FILE_END") or a symbol containing the name of a label or the value "CONTINUE".
Data may be read from a file into a symbol using the corresponding "READ
file symbol" command, where file is the name of the file containing
the data. Attempting to read past the end of the file will cause THINK to execute
an implicit GOTO and jump to the FILE_END location. This may take the same range
of values as the FILE_ERROR and ON_ERROR locations.
READ mols.lis MOLNAME
LET TEXT = "Current molecule is " . MOLNAME
WRITE CONSOLE TEXT
1.5.6 Intrinsic Functions
THINK includes a variety of numerical, string and system instrinsic functions which
are prefixed by "$". Values returned by intrinsic functions can be used like symbols.
Each function takes a number of arguments and returns a single
integer, real or string value that may be assigned to a variable.
In the table below, ival indicates an integer argument, rval
a real argument and cval a character argument.
Function
Return value
Description
$SQRT(rval)
Real
Returns the square root of rval
$EXP(rval)
Real
Returns erval
$LOG(rval)
Real
Takes the natural logarithm of rval
$LOG10(rval)
Real
Takes the common logarithm of rval
$ABS(rval)
Real
Returns the absolute value of rval
$INT(rval)
Integer
Returns the integer part of rval truncated towards zero,
ie $INT(3.4) returns 3, $INT(-3.4) returns -3
$NINT(rval)
Integer
Returns the nearest integer to rval.
If rval>0 $NINT(rval) has the value $INT(rval+0.5).
If rval≤0 $NINT(rval) has the value $INT(rval-0.5)
$CEILING(rval)
Integer
Returns the nearest integer that is greater than or equal to rval,
ie $CEILING(3.1) returns 4, $CEILING(-3.1) returns -3
$FLOOR(rval)
Integer
Returns the nearest integer that is less than or equal to rval,
ie $FLOOR(6.3) returns 6, $FLOOR(-6.3) returns -7
$TRUNCATE(rval1,ival2)
Real or integer
Truncates rval1 to ival2 decimal places, ie $TRUNCATE(2.468,2)
returns 2.46. ival2 must be in the range 0-3. If ival2=0, $TRUNCATE
returns the same value as $INT(rval1)
$ROUND(rval1,ival2)
Real or integer
Rounds rval1 to ival2 decimal places, ie $ROUND(2.468,2)
returns 2.47. ival2 must be in the range 0-3. If ival2=0, $ROUND
returns the same value as $NINT(rval1)
$MAX(rval1,rval2)
Real
Returns the larger value of rval1 and rval2
$MIN(rval1,rval2)
Real
Returns the smaller value of rval1 and rval2
$CPUTIME()
Real
Returns the number of seconds of CPU time used by the current
THINK session
$ICHAR(cval)
Integer
Returns the ASCII value of the first character in cval
$CHAR(ival)
Character
Returns the character corresponding to the ASCII code ival
$INDEX(cval1,cval2)
Integer
Returns the starting position of substring cval2 within
character string cval1. A value of 0 is returned if cval2 is not
found
$LENGTH(cval)
Integer
Returns the length of character string cval
$TRIM(cval)
Character
Returns the string cval with all leading and trailing spaces
removed
$LOWCASE(cval)
Character
Returns the string cval with all uppercase characters converted
to their lowercase equivalents
$UPCASE(cval)
Character
Returns the string cval with all lowercase characters converted
to their uppercase equivalents
$VERSION()
Character
Returns the THINK version number (eg 1.23b)
$FIELD(cval1,cval2)
Real
Returns the value stored in data field cval1 for molecule
cval2
$MOLECULE(ival)
Character
Returns the name of the ival'th molecule within THINK
$ATOM(ival)
Character
Returns the name of the ival'th atom within THINK
$QUERY()
Character
Returns the name of the query molecule
$SITE(ival,protein)
Character
Returns the name of the ival'th site for the specified protein molecule
$KEY(molecule,number)
Integer
Returns the key number if the functional group key is set for molecule or -1 if not set
$SUBSTRUCTURE(molecule,smiles)
Integer
Returns the number of occurences of the substructure defined by the smiles string in the
specified molecule
$FEXIST(cval)
Character
Returns TRUE if the file cval exists, otherwise returns
FALSE
$FSIZE(cval)
Integer
Returns the size of the file cval exists, otherwise returns
-1
$FPATH(ival,file)
Character
Returns the full path to the file when ival is zero or the extension for ival=1; the name for ival=2; the directory for ival=4 or combinations thereof fpr 3, 5, 6, or 7.
$FDELETE(cval)
Character
Attempts to delete the file cval and returns TRUE if the
file is deleted successfully, otherwise returns FALSE
An extension to the SMILES format allows reactions to be stored with a ">" character separating the reactant(s) from the product(s). When reading
proteins, hydrogen atoms are not automatically added whereas by default for small molecules hydrogens
are automatically added to fill incomplete valencies.
Normally SMILES
files would be used for 2D molecules, SD files for 3D molecules and PDB files
for peptides or proteins. Hydrogens will be written for 3D files but not 2D formats.
Atoms are assigned atom types, serial and groups numbers, names etc when the file is processed.
To complete valencies, hydrogens are added to small molecules but not proteins.
If the file does not contain 2D or 3D coordinates THINK will attempt to generate these when required.
Only protein atoms are organised into residues.
2.1 Reading Molecules
The Open node may be used to read molecule data files in the standard formats. The output table includes
columns for the SDF and SMILES formats as well as the data fields. In most cases, other nodes which
read SDF or SMILES format may be used.
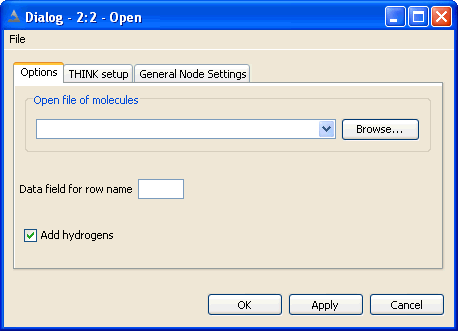
The configuration option dialog is used to specify the filename and control
Whether hydrogens are automatically added (undesirable for substructure search queries).
The name of the data field in a MACCS to be used for the molecule name.
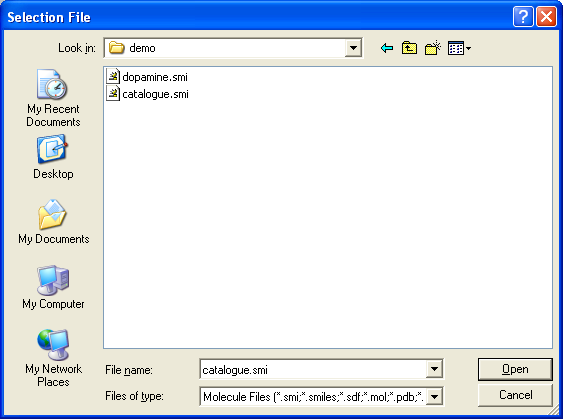
Molecules are usually read using the File > Open dialog, or
the file explorer (see below). Under Windows files may also be dragged from the Windows Explorer
and dropped in the Console window. The dialog does not include functionality to read a subset of molecules
from a file. The option to disable automatic hydrogen addition can be important when reading fragments for substructure
searching and is found on the File Explorer dialog.
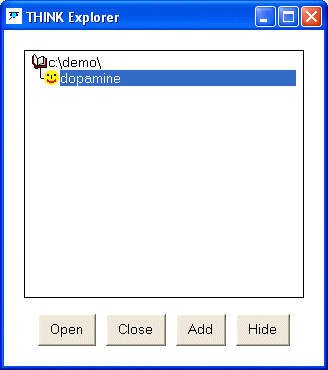
The file explorer contains a list of all SMILES, PDB and SD files that have
been opened by THINK displayed in lower case. It maintains a hierarchy of these files so that the output from searches are
located under the file which was searched with a default name derived from the query.
If the user wishes to read a new file using the explorer,
it must first be added to the list of recognised files by using the Add button.
Once the file is visible in the list it can be opened by double-picking the file,
or by picking Open from the pop-up menu displayed by the right mouse button.
A file may be "closed" when all the molecules that have been read
from that file automatically deleted from THINK (but not from the file on disk),
by picking Close from the right mouse button pop-up menu.
Commands
Dialogs
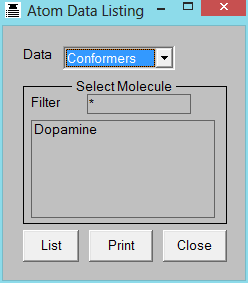
OPEN FILE=dopamine.smi
File > Open
OPEN FILE=dopamine.smi
Select filename from File Explorer then Open
CLOSE FILE=dopamine
Select filename from File Explorer then Close
Note: In THINK v1.25 the file path and extension is omitted from the close command.
Molecules are read through the OPEN command. This command provides the maximum flexibility
- the other routes read all the molecules in the file in a single operation.
When using the OPEN command to read SMILES, PDB or SD files, the user has the
option to read selected molecules from the file through the MOLECULE keyword,
using the molecule names or positions within the file to identify the desired
molecules.
The molecule position is specified through the
keyword construct "MOLECULE=#n" where n is 1 for the first
molecule, 2 for the second, etc. If the file contains molecule names then either
the name ("MOLECULE=name") or the position may be used to identify
the molecule. Several molecules may be read in a single operation if name includes
wildcard characters.
The capability to suppress hydrogen addition is important when reading queries for substructure
searching. The OPEN command allows the user to override the
default setting through the "OPTIONS=NOHYDROGENS" and "OPTIONS=HYDROGENS"
keywords respectively. When automatic hydrogen addition is suppressed, only hydrogen
atoms that are explicitly included in the file will appear in the molecule. Any
hydrogens that would normally be added as a result of THINK interpreting elements
of the form [CH] or [CHn] in SMILES files or interpreting the hydrogen-count
field in SD files are omitted.
Commands
Dialogs
OPEN FILE=dopamine.smi
File > Open
OPEN FILE=capsaicin.smi MOLECULE=#5
Selective read not supported
OPEN FILE=dopamine.sdf MOLECULE=DOPAMINE(1)
Selective read not supported
OPEN FILE=dopamine.sdf MOLECULE=DOPAMINE(1%)
Selective read not supported
OPEN MOLECULE=dopamine.smi OPTIONS=NOHYDROGENS
File explorer option
File names that include spaces may be used, providing they are enclosed
in double quotes "".
Names for the molecules in the file are deduced as follows:
SMILES files: the record contains the field NAME=xxx, or the SMILES
string is followed by the name of the molecule without any preceding "field="
c1ccccc1 ... NAME=BENZENE ...
c1ccccc1 BENZENE ...
SD files: the name is contained in the first or second record of the molecule's
block within the file
PDB files: the name is contained in the non-standard NAME record
at the beginning of the molecule's block within the file. If the file does not contain NAME records,
the name is taken from the MOLECULE:, SYNONYM or EC: values in the COMPND record,
or from the protein ID in the HEADER record. Ligand names are taken from the HET,
HETNAM or HETSYM records.
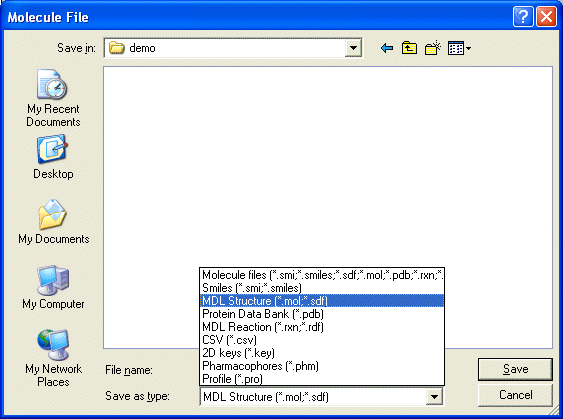
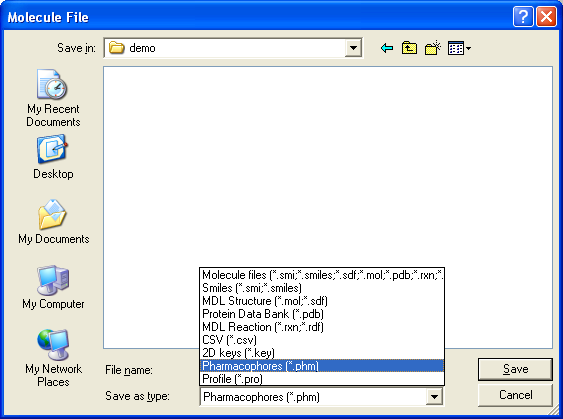
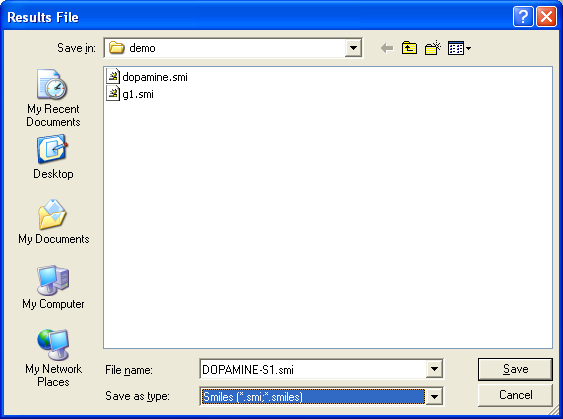
2.2 Saving Molecules
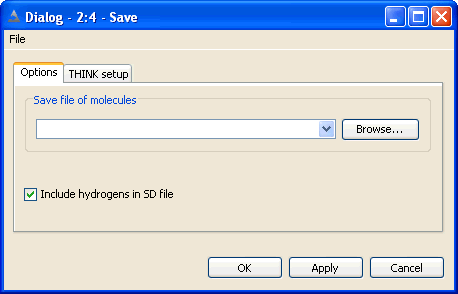
The Save node may be used to write molecule data files in the standard formats. The numerica field
data is included in SD and SMILES files. In most cases, other nodes which write SDF or SMILES format may be used.
The configuration option dialog is used to specify the filename and control
whether hydrogens are included in SD files.
Molecules may be used using the File Save As dialog which saves
all molecules loaded within THINK. It is possible to save a subset of molecules using
the popup menu in a spreadsheet or tile display.
Commands
Dialogs
SAVE FILE=dopamine.sdf
File > Save
SAVE FILE=dopamines.sdf MOLECULES=@SELECTED
Use popup menu in spreadsheet or tile display
Molecules may be saved via the SAVE command.
The "MOLECULE=name" keyword allows specified molecules to be saved using wildcards
and/or a comma separated list of molecule names.
By default any field information associated with the molecules will be written to the SMILES or SD file; this may
be suppressed through the "OPTION=NOFIELDS" keyword.
If the user wishes to generate
and save conformers the "FORMAT=CONFORMERS" keyword must be specified.
THINK provides the option to reduce the number of molecules saved to a file using the "OPTION=FILTER" keyword
to omit those that contain undesirable substructures or property values. This
option would normally be used when saving the enumerated molecules from a combinatorial
chemistry library (see Chapter 15), but may be used when
saving any set of molecules. The substructure and property value filters are taken
from a learn file created by an earlier data analysis calculation (see
Chapter 11). The name of the learn file is automatically
taken from the name of the field that contains activity data (set through the
CUSTOMISE command using the ACTIVITY keyword). If the activity field is not set
then the file "default.lrn" in the THINK_EXEC directory will be used.
Commands
Dialogs
SAVE FILE=dopamine.sdf
File > Save
SAVE FORMAT=CONFORMERS FILE=dopamine.sdf
Conformer save not supported
SAVE FILE=dopamines.sdf MOLECULES=S1,S2,S6,S7*
Specific save not supported
SAVE FILE=dopamines.sdf MOLECULES=@SELECTED
Use popup menu in spreadsheet or tile display
CUSTOMISE ACTIVITY=LOGK
SAVE FILE=LIB1.SMI OPTIONS=FILTER,NOFIELDS
Options not supported
Usage Mode:
3 Creating, Modifying and Deleting Atoms and Molecules
3.1 Creating Molecules
Molecules are normally read into THINK from an external file (see
section 2.1). THINK does not provide a KNIME node for
creating new molecules.
Although molecules are normally read into THINK from an external file (see
section 2.1), there are occasions when it may
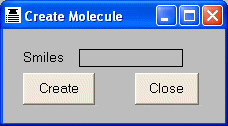
be more convenient to create a simple molecule by typing the appropriate SMILES
string instead of creating a file and then reading the file into THINK. Molecules
may be entered by typing the SMILES string into the Edit > Create dialog
and then picking the Create button.
Commands
Dialogs
OPEN FILE=TTY MOLECULE=c1cncc1
Edit > Create
Although molecules are normally read into THINK from an external file (see
section 2.1), there are occasions when it may
be more convenient to create a simple molecule by typing the appropriate SMILES
string instead of creating a file and then reading the file into THINK. Molecules
may be entered by using the OPEN command and supplying "TTY" as the name of the input
file. The SMILES string is specified using the MOLECULE keyword.
Note that the filename is case-sensitive (to support the LINUX operating
system) and "TTY" must be supplied in uppercase.
Commands
Dialogs
OPEN FILE=TTY MOLECULE=c1cncc1
Edit > Create
3.2 Editing Molecules
The display node includes functionality to edit a molecule in 2D using the mouse, the drawing tools
and the right mouse button popup menu. The drawing tools provide a subset of the functionality found on
the menu and are described in the following table.
The 2D molecule display includes functionality to edit a molecule using the mouse, the drawing tools
and the right mouse button popup menu. The drawing tools provide a subset of the functionality found on
the menu and are described in the following table.
Tool
Menu item
Action
Element ..
Selects the element for new or existing picked atoms (includes a periodic table)
This is also used to specify R-groups for combinatorial chemistry reaction schemes and other wildcard queries.
Insert atom
Insert atom at picked coordinates
Insert bond
Insert bond between picked atoms
Bond order ..
Changes the order of the picked bond
Sprout atom
Insert atom connected to picked atom
Delete atom
Delete picked atom
Delete bond
Delete bond between picked atoms or bond
Rings > 6 atoms
Insert 6-membered saturated ring at picked coordinates, atom or bond
Rings > 5 atoms
Insert 5-membered saturated ring at picked coordinates, atom or bond
Rings > 4 atoms
Insert 4-membered saturated ring at picked coordinates, atom or bond
Rings > 3 atoms
Insert 3-membered saturated ring at picked coordinates, atom or bond
Rings > Aromatic 6
Insert 6-membered aromatic ring at picked coordinates, atom or bond
Rings > Aromatic 5
Insert 5-membered aromatic ring at picked coordinates, atom or bond
Undo
Undo previous action (maximum 10)
Flip/Rotate
Flips molecule about X or Y axes and rotates about Z axes
Tidy
Regenerates 2D coordinates
Save
Saves molecule
Notes
When a bond is picked with the Insert Bond tool the bond order cycles through the series single > double > triple.
When an atom is picked with a ring insertion tool, the atom is included in the ring.
When a bond is picked with a ring insertion tool, the bond is included in the ring. This can be used
to build fused rings. The order of the existing bond will be changed to aromatic when an aromatic ring is
being inserted.
Hydrogens are not normally displayed and need not be drawn - they are added automatically when using
other functionality in the program.
The delete tool can be combined with the shift key to delete a fragment by clicking an atom or a chain by clicking a bond.
A reaction drawing mode can be enable/disabled using the right mouse button popup menu.
The text command mode provides some functionality to modify or delete existing atoms and bonds but does
not offer functionality to create new atoms.
Bonds may be made or broken within the MODIFY command using the MAKE-BOND and
BREAK-BOND keywords respectively. The bond is defined by the two atoms and the
bond order:
Symbol
Bond Order
-
single
:
aromatic
=
double
#
triple
The MAKE-BOND keyword may be used to change the bond order of an existing bond.
The atom type, name, serial number or group number of any atom or collection
of atoms may be changed using the MODIFY command. The atom(s) to be altered are
identified with the CHANGE keyword, and the new data is supplied with the TYPE,
NAME, SERIAL and GROUP keywords. If a set of atoms is to be changed, it may be
specified as a range of serial numbers, such as (3:7), or via a symbol (see sections
1.4 and
1.5.1). All the
atoms in the set will be given the new name, etc, which may lead to multiple atoms
sharing the same identification, so this option must be used with caution.
If the keyword construct "SERIAL=#" is used, the serial number of
each atom in the set will be altered to reflect the position of the atom within
the molecule. Thus, the first atom will be given serial number 1, the second 2,
etc. When applied to all the atoms in a molecule, this is a quick method of assigning
unique serial numbers.
Atoms may be deleted using the "DELETE=atoms" keyword. After deleting atoms or changing the bonds or atom types within
a molecule, it is recommended that the molecule is rebuilt to update the coordinates
and connectivity. THINK will regenerate the 2D or 3D coordinates or just the connectivity
depending upon the REBUILD option used (2D, 3D or CONNECTIONS respectively). The
molecule to be rebuilt is identified with the MOLECULE keyword.
The molecule name may be altered. The molecule is identified with the MOLECULE
keyword and the new name is supplied with the TO keyword. If desired, the conformation
number may be set or altered as part of the same command.
THINK does not include a node to delete a molecule. However, the standard KNIME filter node allows
rows (molecules) to be eliminated. Some THINK nodes have the option to process just the first molecule
which often eliminates the need to use the filter node.
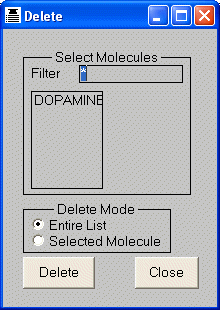
The Edit > Delete dialog is used to delete molecules. An individual molecule
may be deleted by selecting its name from the list, or all the molecules in the
list may be cleared in a single operation. Selective lists of molecules may be
deleted by changing the filter at the top of the dialog (the default filter
is "*", which lists all molecules) and then deleting all the molecules
in the resulting list.
Molecules may be deleted using the DELETE command. The molecule(s)
are specified with the MOLECULE keyword, and may include wildcards. The keyword
construct "MOLECULE=*" may be used to delete all molecules.
Individual atoms or sets of atoms may be deleted using the DELETE keyword within
the MODIFY command.
Commands
Dialogs
DELETE MOLECULE=CAPSINV5
Edit > Delete
DELETE MOLECULE=GLY*
Edit > Delete
Usage Mode:
4 Visualisation
Although there are other third party nodes which can be used to display molecules, most of these do not
provide the functionality required to view a series of super-imposed molecules for instance docked
into a protein.
In THINK 1.42, the display node is only supported under Windows.
Click on the box to display the corresponding dialog.
Within THINK both the molecules and their data may be visualised in a variety
of different ways. The term visualisation incorporates simple data listings
(eg lists of torsion angles) as well as molecular properties (eg lipophilicity).
4.1 Input Molecules
The input table is a set of molecules with a SDF column with 2D or 3D coordinates or a SMILES column. Implicit
inputs are associated with previously executed Search, Similarity or Docking nodes. THINK will automatically
generate 2D or 3D coordinates if they are required for the display mode and absent from the input table.
Before any molecule can be visualised it must be read into THINK for instance from a SMILES,
PDB or SD file. If the file contains several molecules, of which only a subset
is required, these may be selectively loaded (see section 2.1).
Alternatively, all the molecules in the file may be read and the desired molecules
specified as part of the visualisation.
Commands
Dialogs
OPEN FILE=capsaicin.smi
File > Open
OPEN FILE=m2.sdf MOLECULE=TC1*
Selective read not supported
OPEN FILE=m2.sdf
File > Open
When a molecule is read from a SMILES or SD file, any hydrogens specified as
explicit atoms in the file will be automatically included in the picture. Hydrogens
that are specified implicitly via a hydrogen-count (ie as [CHn] in a SMILES
file or through the hydrogen-count flag in a SD file) are excluded from the picture,
as are any hydrogen atoms that are added automatically to complete the valencies.
THINK will automatically generate the 2D or 3D coordinates in order to display
the molecules if the necessary data was not read from the SMILES or SD file.
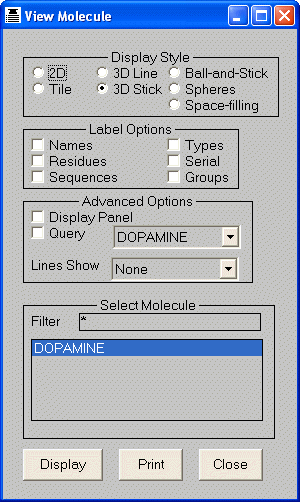
4.2 Viewing Molecules
The Display node provides the capability of viewing single or multiple molecules in 2D and 3D. If more
than one molecule is present, the 2D display is tiled and edit functionality is not available. In 3D multiple
molecules can be displayed superimposed on each other or docked into a protein. The output is the same
as the input table of molecules, unless the Save option is used when only the selected molecules are
included in the output table.
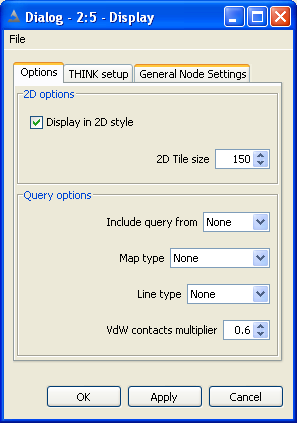
The configuration option dialog is used to select 2D or 3D display mode
and control
The 2D tile size (in pixels) for each molecule.
Whether to include a query molecule and if so select the query for the most recently executed Search, Similarity or Docking nodes.
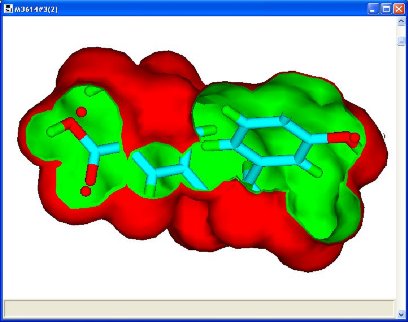
An optional volume map display associated with a pharmacophore query (created by a previously executed pharmacophore
node); volume or potential map for the query molecule or a union volume map for all the molecules also
known as a ligand map.
The line type for showing correspondence or interactions between a protein query and docked ligands.
The contacts to VdW multiple for scaling down interactions (leaving the most significant).
Molecules may be displayed in various 3D styles, using a 2D representation with one molecule per
screen, or with a tiled display showing many molecules on a single screen.
Molecules may be displayed in various 3D styles, using a 2D representation with one molecule per
screen, or with a tiled display showing many molecules on a single screen.
Pictures may be displayed on the screen or printed on the default printer using
the DISPLAY and PLOT commands respectively. To avoid producing large quantities
of output on the printer, it is recommended that the conformer display is avoided
and that molecules are printed selectively when more than one or two are present
(unless otherwise specified, all molecules will be displayed or printed).
4.2.1 2D Display Mode
Molecules may be displayed using a 2D representation with one molecule per
screen, or with a tiled display showing many molecules on a single screen. The
tiling is automatically adjusted when the window size is altered. The vertical
scroll bar and scrolling arrows allow the user to step through the molecules,
displaying each in turn. To avoid clutter, dummy atoms are automatically omitted
from the picture when a small window is used or in the tiled display style.
When a query molecule is included, the corresponding substructures are highlighted.
Molecules in the tile display may be selected by clicking on them. A pop-up menu permits further
manipulations of the selection.
Item
Description
Save Selected
The selected molecules are saved
Invert Selection
The select subset and unselected subset are swapped
Select Similar
A similarity search is performed to select similar molecules
Select Family
De Novo derivatives generated from the same original molecule are selected
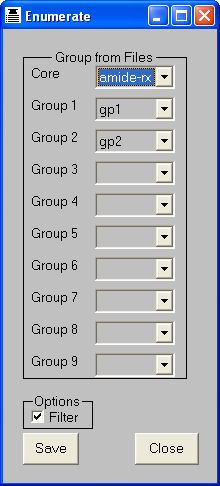
If an R-group (see Chapter 15) is displayed in 2D,
the explicit connection atoms are represented by the digits 1-9, corresponding
to the atom types of the atoms, whilst generic connection atoms are represented
by "*".
Commands
Dialogs
Output
DISPLAY MODE=TILE
View > Molecule > Tile
4.2.2 3D Display Mode
When molecules are displayed in 3D different colours are assigned to represent the elements
with each molecule. As with 2D representations, the vertical scroll
bar and scroll arrows may be used to step through the molecules. When a query molecule is specified, it is also displayed.
The default scaling is for the query (if specified) or the last molecule.
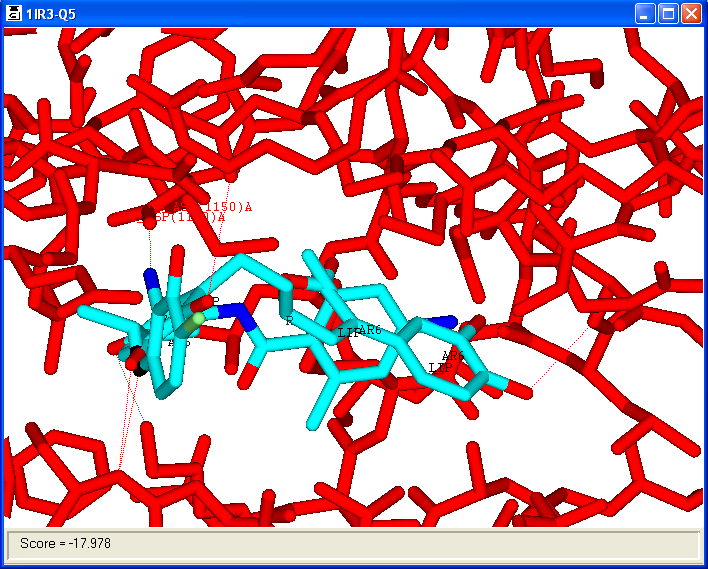
View > Molecule > 3D Stick; Display Panel; Query:1IR3
PLOT MODE=3D
View > Molecule > Print
DISPLAY MODE=CONFORMERS
View > Conformers
The Display Panel can be very useful for reviewing hits from 3D/SITE searches
providing full control of which molecules are displayed, their colour etc.
In the Display Panel the list of constituent atoms is shown vertically and the molecules horizontally.
Atoms can be selected by clicking on the list with the left mouse button optionally in combination with the SHIFT
or CONTROL keys to select sets of atoms. By default, selecting constituent atoms causes a molecule to
be displayed and the selected atoms to be coloured black. The tools at the top of the Display Panel allow
control of the colours and visibility of atoms. Molecule colour mode provides a different colour for
each molecule and is useful when displaying multiple molecules. The popup menu selected with the right mouse
button provides further display options as outlined in the following table.
Item
Description
Select Whole molecule
Selects all the atoms in the molecule clicked
Select Chain
Selects the atoms in the chain of the clicked atom (proteins only)
Select Site
Selects the atoms in the site of the clicked atom (proteins only)
Invert Atom Selection
For the clicked molecule switch selected and unselected atoms
Colour by
Molecule
Colour the selected atoms using the molecule colour
Atom type
Colour the selected atoms by standard atom colours
Residue type
Colour the selected atoms according to residue classification (proteins only) Hydrophobic: Red; Hydrophilic: Green
Hydrophobicity
Colour the selected atoms by lipophilicity Lipophilic: Red; Hydrophilic: Green
Charge
Colour the selected atoms according to residue charge (proteins only) Positive: Green; Neutral: Molecule colour; Negative:Red
Hide
Atom Selection
Disables display of selected atoms
Side-chains
Disables display of atoms in selected side-chains (proteins only)
Hydrogens
Disables display of hydrogens in selected atoms
Dummies
Disables display of dummy atoms (rings centroids etc) in selected atoms
This molecule
Disables display of all atoms in clicked molecule
All molecules
Disables display of all molecules
Show
Atom Selection
Enables display of selected atoms
This molecule
Enables display of all atoms in clicked molecule
All molecules
Enables display of all molecules
The style of a 3D picture may be changed using the pop-up menu that is displayed
when the right-hand mouse button is clicked on that window. Display styles available include
stick, ball-and-stick and space-filling representations.
4.2.3 3D Manipulation
3D pictures may be manipulated interactively using the mouse in conjunction
with the pop-up menu (displayed when the right-hand mouse button is clicked).
The type of manipulation (X-Y rotation, X-Y translation, scale etc) is selected
from the menu and picture manipulated by moving the mouse with the
left-hand button depressed. It is possible to avoid selecting from the menu by
using other mouse buttons or keyboard keys as indicated in the following table
which lists the items on the menu.
Reducing the z-depth by moving the mouse down increases the contrast between the front and
back of the molecule(s) and ultimately clips the front and back.
Motion
Key
Mouse button
X-Y Rotate
Control
Middle
X-Y Translate
Shift
Right
Z Rotate
Z (X-direction)
Z Translate
Z (Y-direction)
Bond-rotation
Space (X-direction)
Scale
Tab (X-direction)
Depth
Tab (Y-direction)
Scroll
Global motion
Toggles between global and fragment motion
Orientate
Centre
Moves to centre on a clicked atom
Bond/Vector
Moves to view along a bond or the line between 2 clicked atoms
Plane
Moves to view on to a plane defined by 3 clicked atoms
Superimpose
Overlaps molecules by clicking 3 pairs of atoms
Reset
Reverses global motion
Map
Toggles visibility of map
Edit Centre
Used to set protein site query options
The bond for rotation is selected by clicking on it when the end nearer the cursor is moved and the number of
degrees rotation is reported to the status bar of the window.
When global motion is disabled, the fragment moved is selected by clicking an atom in it.
The Edit > Rotate dialog is used to manipulate molecules without displaying them.
A global rotation takes all the
atoms and applies a rotation about the X, Y or Z axis through the coordinate origin.
Note that in the dialog, the rotation
takes place as soon as the angle is entered into X, Y or Z box.
Bond rotations may also be performed through the Edit > Rotate dialog. The
two connected atoms are specified as Atom 1 and Atom 2, and the angle of rotation
is supplied in degrees. All the atoms connected to Atom 1 (except through Atom
2) will be moved.
The simplest keyboard manipulation is a global rotation, which takes all the
atoms and applies a rotation about the X, Y or Z axis through the coordinate origin.
This is achieved with the ROTATE command using the ABOUT
and ANGLE keywords to define the axis and angle (in degrees) respectively.
Bond rotations are achieved using the "ABOUT=atom1-atom2" keyword construct,
when all the atoms connected to atom1 (except throught Atom 2) will be rotate by the number of degrees specified with
the ANGLE keyword.
The ROTATE command may also be used to orientate a molecule along the line between two atoms (such as a bond); on to
a plane defined by 3 or more atoms; to superimpose one molecule on another by 3 or more pairs of atoms; and
to orientate maps to their best plane projections.
Commands
Dialogs
ROTATE ABOUT=Y ANGLE=45
Edit > Rotate
ROTATE ABOUT=(2)^ASP-(5)^ASP ANGLE=90
Edit > Rotate
4.2.4 Annotation and Labelling
Any 2D or 3D display may be annotated by labelling the atoms within the molecule,
although this is not recommended with the tiled display because it produces a
cluttered picture. THINK provides options to label the molecules with their:
Atom type
Serial number
Group number
Atom name
Residue name
Sequence number, insertion code and chain id
Placing the cursor over a dummy atom in the picture (AR5, AR6 or LIP) will
automatically label all the atoms that contribute to that dummy atom. Distances,
angles and torsion angles may be measured by picking atoms from a 3D picture -
the results are displayed in the bar at the bottom of the picture window.
3D pictures can be annotated to show the CPK or VdW inter-atomic contacts,
using dashed lines to connect each pair of atoms that are in contact.
The type of contacts shown (CPK or VdW) is controlled though the CUSTOMISE CONTACTS setting.
3D pictures can be annotated to show the CPK or VdW inter-atomic contacts,
using dashed lines to connect each pair of atoms that are in contact.
If there are two or more molecules present, and one is defined as the query,
3D pictures can also be annotated to show the interactions or the atom correspondence
(mapping) between the query and the other molecules. The interactions are a subset
of the contacts between the two molecules: only contacts between atoms that are
complementary pairs of interaction centres are included (see the THINK
Theory Manual for details). The atom mapping between the query and a second
molecule shows atoms which have identical serial numbers. It can be used to show
the correspondence between atoms in a 3D query and a hit found during a search,
or between atoms in a site query and the originating protein atoms.
Use of inter-atomic or inter-molecular annotations disables the automatic labelling
of dummy atoms and their defining atoms.
The functional group key or fingerprint indicates the presence of absence of various
functional groups within each molecule. The Functional Group node does not provide a view option
in THINK 1.42.
The configuration option dialog
controls whether the output keys are stored as HEX in a single column or in separate columns. This data
can be processed by standard KNIME nodes.
For each molecule loaded, THINK will automatically calculate a functional group
key, or fingerprint, indicating the presence or absence of various functional
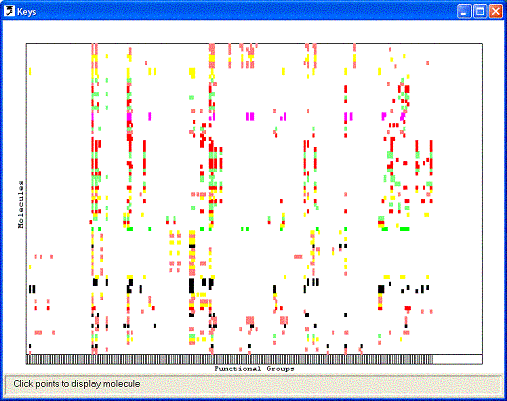
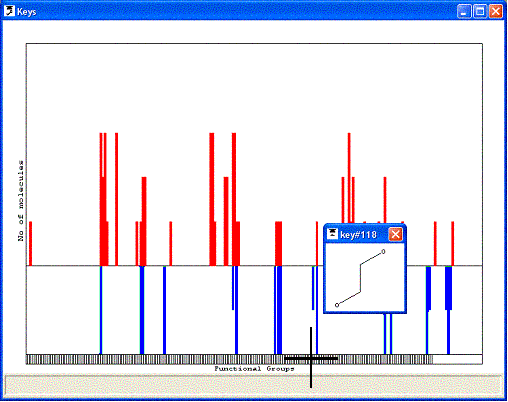
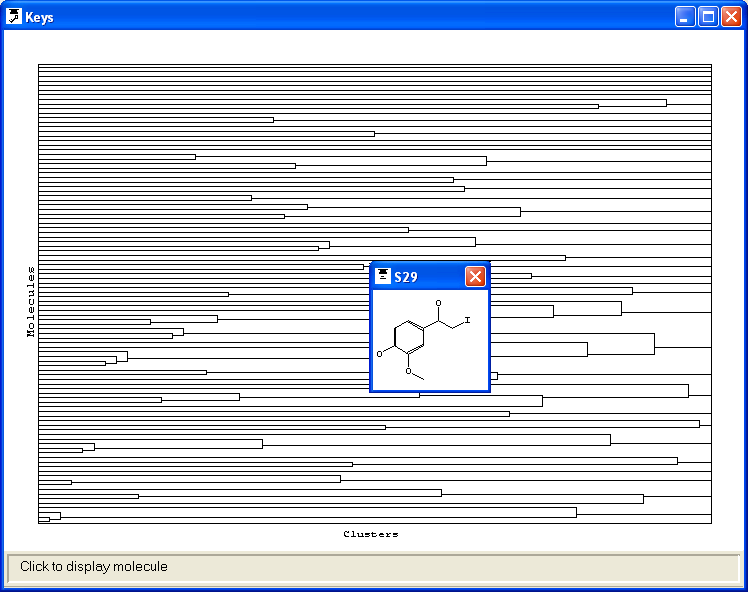
groups within that molecule. The keys can then be displayed as a 2D key plot:
each molecule is represented by a row on the plot and each functional group by
a column. If a particular functional group is present in a molecule, a point is
placed the intersection of the appropriate row and column. There is insufficient
room on the plot to show the molecules explicitly, but picking any point on the
plot will pop-up a 2D representation of the associated molecule. The functional
group associated with any column on the plot can be displayed in a pop-up window
by picking the box on the horizontal bar across the bottom of the plot.
The rows on the 2D key plot may be colour-coded to show the activity of each
molecule. The activity data is taken from a field that was loaded when the molecules
were read from the SMILES or SD file; the user must specify the name of this field.
Commands
Dialogs
Output
DISPLAY MODE=KEYS ACTIVITY=EC50
View > Keys
PLOT MODE=KEYS
View > Keys
An alternative way of visualising the functional group keys is to display them
as a histogram showing the number of molecules that contain each functional group.
The histogram may also be used to show the number of times each functional group
occurs in highly active or inactive molecules (ie to ignore molecules that lie
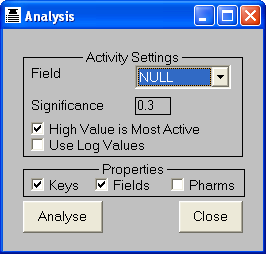
in the middle of the activity range). To achieve this, the user needs to supply
the name of the field containing the activity data, and a significance value in
the range 0-0.5. Molecules whose activities lie within this fraction of the top
or bottom of the activity range are included in the histogram. If a significance
value of 0.5 is specified then all molecules will be included. The activity-coded
histogram is drawn with the numbers of active molecules above the origin line
and inactive molecules below the line, using the appropriate colours for active
and inactive molecules.
For each molecule loaded, THINK can calculate a functional group
key, or fingerprint, indicating the presence or absence of various functional
groups within that molecule.
Commands
Dialogs
KEY MOLECULE=*
Automatically calculated
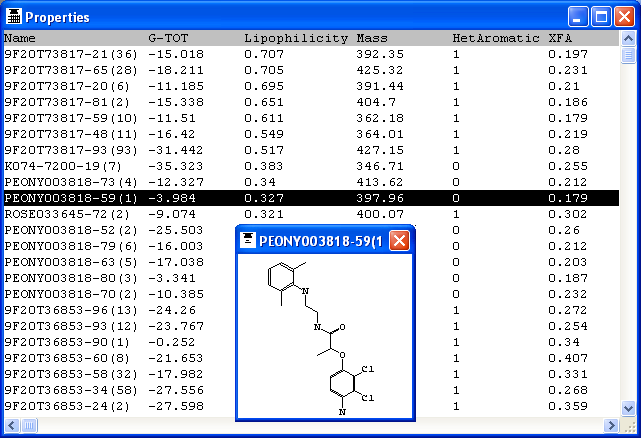
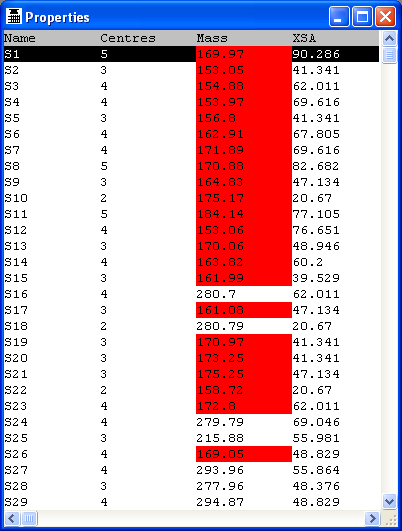
4.4 Molecular Properties
The Properties node calculates a set of 2D and 3D properties and returns these with the
molecular structures in the output table. This table may be used as input to standard KNIME nodes.
There is no Options dialog.
THINK will evaluate or calculate a set of 2D and 3D molecular properties for
the current molecules. These values, along with the contents of any data fields
loaded from the SMILES or SD file (external data fields) may be viewed in a spreadsheet.
Normally, all molecules within THINK would be included in the spreadsheet, but
a subset of molecules may be used.
THINK will evaluate or calculate a set of 2D and 3D molecular properties for
the current molecules. These values, along with the contents of any data fields
loaded from the SMILES or SD file (external data fields) may be listed or exported.
Once calculated, the rows in the spreadsheet may be reordered by sorting the
contents of a column into ascending or descending order, or rows may be deleted.
Columns may be deleted or reinserted (inserting a column that is already present
merely changes its position in the spreadsheet). All these operations are available
through the pop-up menu displayed when a column is picked with the right mouse
button. As an alternative to using the pop-up menu, a column may be sorted by
picking the column title once or twice (the second pick reverses the sorting order)
and may be moved to a new location in the spreadsheet by dragging the column title.
Picking the name of a molecule from the first column will pop-up a 2D representation
of that molecule and will also update the picture window (if present) to show
the same molecule. If a 3D display window is open, the contents of the window are changed
to the molecule(s) that have been selected. Multiple molecules can be selected by holding down the SHIFT
key when picking to select a range. Holding down the CONTROL key allows molecules to be toggled in and
out of the selection. The pop-up menu has functionality to
Save the selected molecules to a named file
Invert the selection
Select similar molecules to the most recently selected molecule
Select molecules in the same family (with the molecule name ending -n where n is a number)
Two of the columns in the spreadsheet may be plotted against each other, using
a third column to colour-code the points representing the molecules. This is done
by setting two columns as the X- and Y-axes of the plot and the third as the "activity"
column (even if it does not really contain activity data) through the right mouse
button pop-up menu. The plot will be drawn as soon as the X- and Y-axes have been
defined. Molecules may also be selected off this plot to be displayed as with the worksheet including
use of the SHIFT and CONTROL keys. In addition, a sphere select centred on a molecule
can be used by moving the mouse while holding the left button down to select molecules in the selected range.
Alternatively a rectangle select can be used when there is no molecule at the coordinates when the left button
is pushed down. Rectange and sphere selections can also be modified using the SHIFT and CONTROL keys.
The same pop-up menu for manipulating selections is available in the plot and the spreadsheet.
If a learn file created by an earlier data analysis calculation (see Chapter
11) is available, the properties of the current molecules
may be compared with those contained within the file. In this case, the spreadsheet
will only contain those properties that occur in the learn file. Values that lie
outside the property ranges taken from the file are highlighted. The filename for the
learn file is derived from the activity field by adding the suffix ".lrn" and must
be present in the working folder.
When reviewing 3D/SITE search results, it can be useful to compare a known ligand and
the docked conformers of that ligand. If the known ligand is specified as the query
then field containing the RMS deviations for all atoms and non-H atoms mapped by serial number
are included. A further field RMS-Match contains the RMS deviations of just those atoms
which matched the original 3D/SITE search query. If the protein is designated as the query
then the contributions to the score (G-TOT) are computed and included in the table.
Commands
Dialogs
Output
LIST INFO=PROPERTIES OUTPUT=WINDOW
View > Table
LIST INFO=PROPERTIES OUTPUT=WINDOW ACTIVITY=ec50
View > Table
Commands
Dialogs
Output
LIST INFO=PROPERTIES
View > Table
4.5 View Diversity
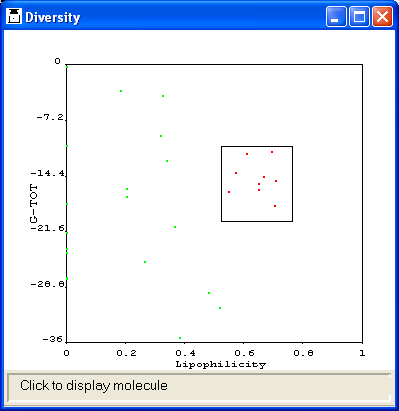
The property diversity of the molecules can be shown in a panel plot or a 3D
cube plot. In the former, the plot is divided into 25 separate tiles or individual
plots, each representing a different number of centres. Within each tile, volume
is plotted along one axis and lipophilicity along the other. In a 3D cube plot,
volume, lipophilicity and the number of centres are plotted along the three axes
of a cube that can be rotated.
If the molecules have associated activity data (read from a field in the SMILES
or SD file), they may be colour-coded in the diversity plot to show their activity.
The user must supply the name of the field containing the activity data.
See Chapter 11 for more information on the calculation
of property diversity
Commands
Dialogs
Output
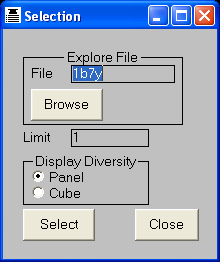
PLOT MODE=PANEL MOLECULE=TC1*
View > Diversity
DISPLAY MODE=CUBE ACTIVITY=EC50
View > Diversity
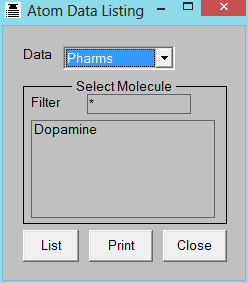
4.6 View Atom Data
THINK can list various types of atomic data to the THINK console window
or to a printer. This data currently includes:
Keywords
Property
TYPES
Atom types
CONNECTIONS
BOND-ORDERS
Connections or bond orders
2D-COORDS
3D-COORDS
2D or 3D coordinates
LENGTHS
ANGLES
TORSIONS
Bond lengths, angles and torsion angles
FLEXIBILITY
Flexibility
CONFORMERS
Torsion angles within acceptable conformers
KEYS
Functional group keys (in hexadecimal)
PHARMACOPHORES
To list the pharmacophores which may be exhibited by a molecule
MAPS
To list the maps currently defined
SITES
To list the binding sites for a protein
PROPERTIES
Molecular properties (2D and 3D) as described below
The listing will be displayed in the THINK console window, or it may be sent
to a printer if "OUTPUT=PRINTER" is specified. At the end of the molecular
property listing, THINK lists the range of each property in the form min:max.
Note that the molecular properties will be listed to the spreadsheet if "OUTPUT=WINDOW"
is specified.
Commands
Dialogs
LIST INFO=TYPES
View > Atom Data > Data:Types; List
LIST INFO=FLEXIBILITY OUTPUT=PRINTER
View > Atom Data > Data:Properties; Print
Usage Mode:
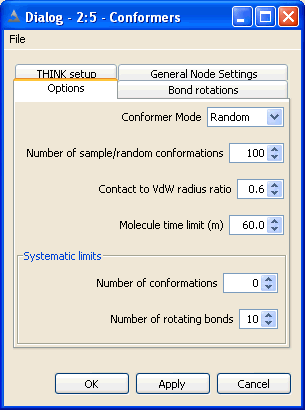
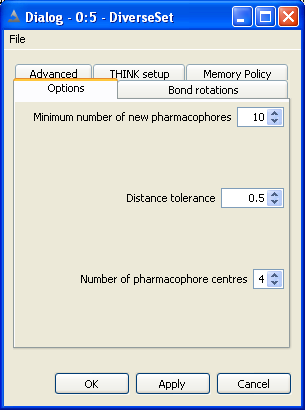
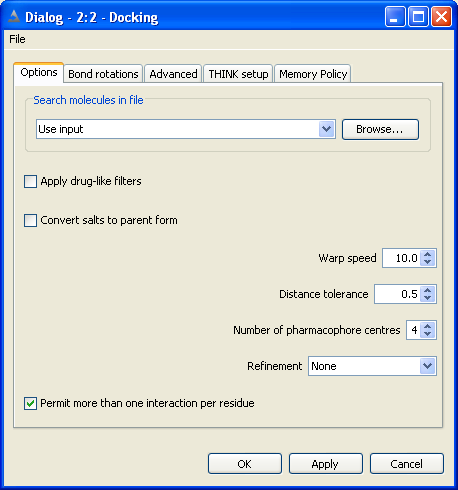
5 Conformer Generation
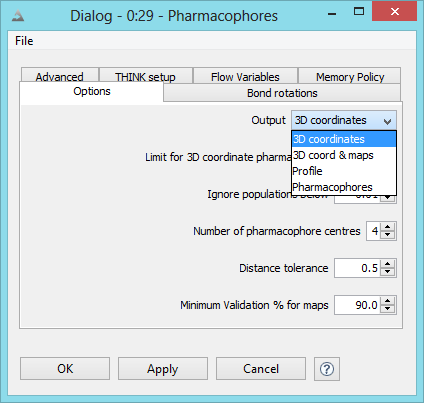
Conformers may be generated for a molecule and exported from the node as a table.
The configuration options dialog is used
to specify the conformer generation mode and when this is not systematic (ie regular or random sampling)
the number of conformers required can be specified. This dialog includes several less frequently used
options which are described below.
The configuration bond rotations dialog is used
to set the rotational increments about the bonds.
It is not necessary to generate conformers prior to performing a 3D search, docking or
generating pharmacophores. THINK will generate them automatically without storing in order to conserve
memory and improve performance.
This chapter serves to describe the use of the conformational generation settings. The conformers
generated may be used by some other software.
Conformer generation is sometimes described as conformer search because
it is searching through the conformational space of the molecule.
Conformers may be generated for a molecule and displayed or saved
to a file. The torsion angles about the rotatable bonds for all acceptable conformers
may be listed to the console window.
It is not necessary to generate conformers prior to performing a 3D search, docking or
generating pharmacophores. THINK will generate them automatically without storing them.
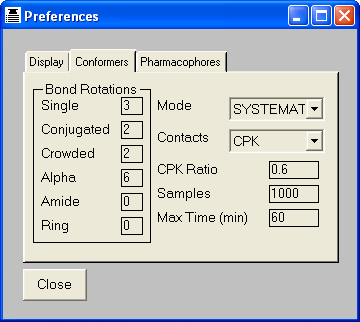
This chapter serves to describe the use of the conformational generation settings.
These settings are independent of the
molecules being processed and the operation being performed (display, listing,
3D searching, etc). They are global settings defined through the customise utility.
Conformer generation is sometimes described as conformer search because
it is searching through the conformational space of the molecule.
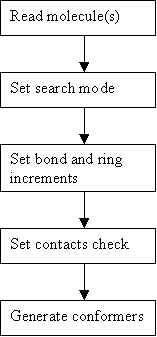
5.1 Input Molecule(s)
The input table is a set of molecules with a SDF column with 2D or 3D coordinates or a SMILES column.
THINK will automatically
generate 2D or 3D coordinates if they are required for the display mode and absent from the input table.
By default, only the first molecule (row) in the table is processed. The option to process all rows is
found on the THINK setup tab.
The molecule or molecules whose conformers are required must be read into THINK
from SMILES or SD files before they can be processed. Molecules may also be
read from PDB files. However, these are normally used to store proteins or peptides
which are frequently too large or too flexible for conformer generation.
The molecule or molecules whose conformers are required must be read into THINK
from SMILES or SD files before they can be processed. If the file contains additional
molecules, the desired subset may be read selectively. Alternatively, the molecules
to be processed may be identified during the calculation. Molecules may also be
read from PDB files. However, these are normally used to store proteins or peptides
which are frequently too large or too flexible for conformer generation.
Commands
Dialogs
OPEN FILE=capsaicin.smi
File > Open
OPEN FILE=m2.sdf MOLECULE=TC1*
Selective read not supported
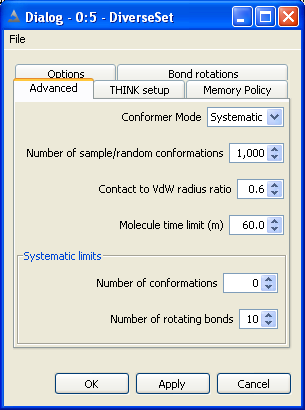
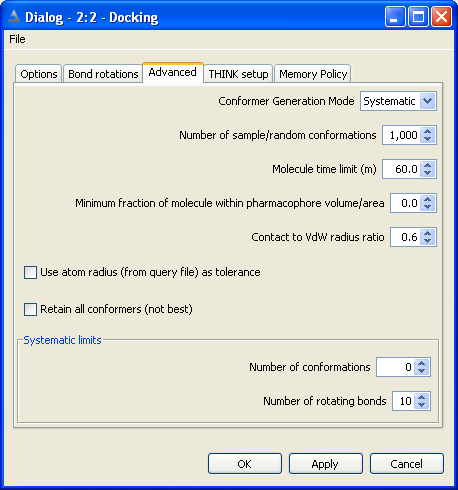
5.2 Set Search Mode
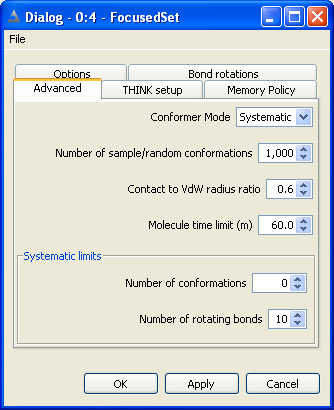
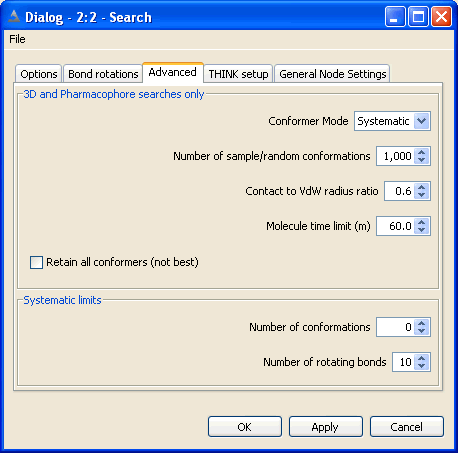
Normally the conformers of a molecule would be generated by systematically
rotating each rotatable bond, and taking each ring increment in turn. This may
generate a large number of conformers, particularly if the molecule is fairly
flexible, and is often quite slow. The process is accelerated by applying a
contacts check (see below) to eliminate conformers with atoms in contact.
An alternative mode of conformer generation that is frequently used with a
very flexible molecule is to generate a random sample of conformers and assume
that these form a representative set of the complete conformational space for
that molecule. However, since the conformers are generated at random, there is
nothing to prevent the same conformer being generated several times.
To overcome this problem and generate a more representative set of conformers,
THINK also provides the option to sample the conformers regularly across the complete
conformational space of the molecule.
Both sample-based methods require the user to supply the number of conformers
required in the sample.
For very flexible molecules especially those with 10 or more rotatable bonds systematic search usually
takes too long. The systematic limits automatically switch to using sampling rather than systematic mode
if the number of rotatable bonds or number of conformers exceeds the systematic limits.
The timeout option allows configures THINK to abort processing of a molecule after the CPU time exceeds the
limit and proceed to the next molecule.
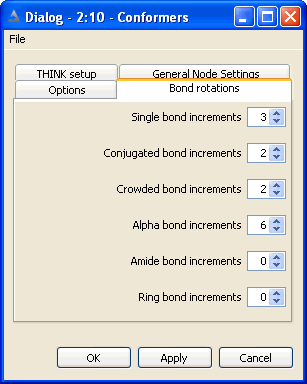
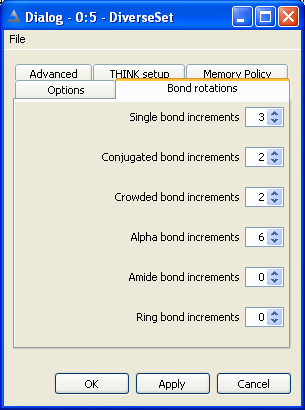
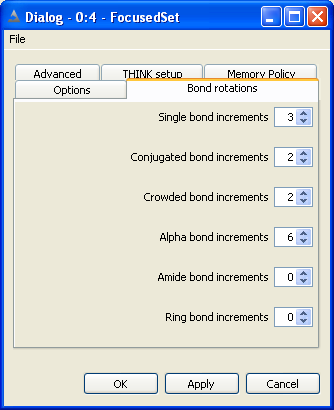
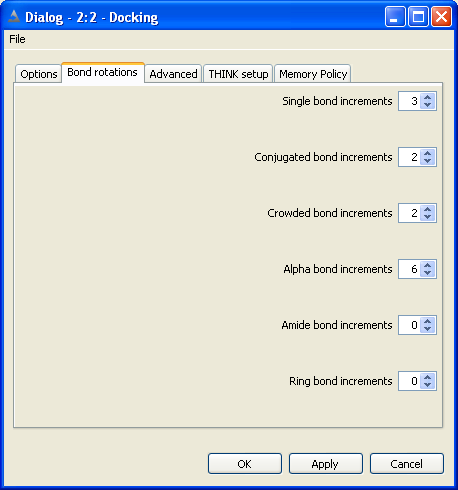
5.3 Set Bond and Ring Increments
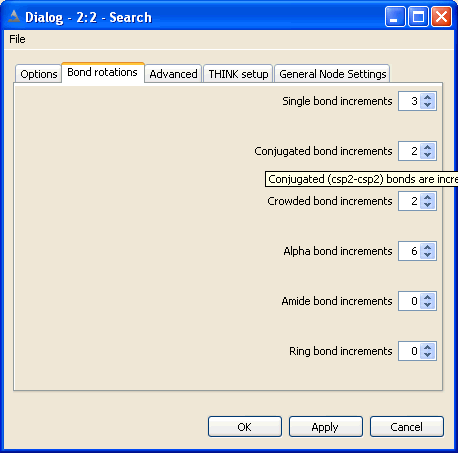
The non-ring single bonds within the molecule are divided into five classes:
Single bonds - between sp3 atoms
Alpha bonds - between sp2 and sp3 atoms
Conjugated and non-conjugated bonds - between sp2 atoms
Crowded bonds - between sp2 atoms which are neither obviously conjugated nor non-conjugated
Amide bonds
The number of points, or increments, to be applied about bonds of the appropriate
type may be set independently for each class of bond. Setting the number of points
to zero will eliminate all bonds of that class from the conformer generation.
Conjugated bonds are normally processed with torsion angles +/-180 while non-conjugated bonds
use +/-90. The crowded classification is used when it is not obvious whether the degree of steric
hinderance is sufficient to cause the bond to be non-conjugated. Crowded bonds are best sampled
at 90, 180, -90 and -180 degrees.
Normally bonds within rings are ignored during the conformational generation.
If they are to be included, the maximum number of points to be sampled about each
ring must be defined. Note that this is the maximum number of ring conformations
to be used, not the number of points about each bond within the ring. There is
currently no distinction between different types of ring: all rings will use the
same number of increments.
The total number of conformers generated may be reduced by applying a contacts
check to eliminate all conformers that contain one or more pairs of atoms in VdW
or CPK contact. This has the additional benefit of speeding up the calculation
since THINK will automatically skip all conformers that do not move the touching
atoms apart.
By default, the ratio of the CPK radius to the VdW radius of an atom is defined
as 0.6, although this value can be changed by the user.
The generated conformers are written to the output table in SDF format. Each
conformer is given a unique name of the form molecule(n) where molecule
is the name of the original molecule and n is an integer number used to
distinguish the conformers.
Conformers are automatically generated as they are displayed, listed or saved
to an SD file. They are also generated implicitly during 3D searches.
Within the SD file, the conformers are saved as separate 3D molecules. Each
conformer is given a unique name of the form molecule(n) where molecule
is the name of the original molecule and n is an integer number used to
distinguish the conformers. There is no option to store conformers in a SMILES
file because the SMILES format does not include coordinate information, and without
coordinates there is no way to re-create the conformers when the file is read.
The conformers are transient - they have no separate existence within THINK
and are discarded as soon as they have been displayed, listed or saved - so the
user is left with the original molecule at the end of the calculation. If the
conformers are required for further analysis, they should be saved to an SD file
and then read back from the file as new molecules.
Commands
Dialogs
DISPLAY MODE=CONFORMERS ...
View > Conformers
LIST INFO=CONFORMERS
View > Atom Data > Data:Conformers
SAVE FILE=dconf.sdf FORMAT=CONFORMERS
Option not supported
Usage Mode:
6 Pharmacophores
The pharmacophores node calculates the pharmacophores for a series of molecules and outputs one of
The 3D coordinates for the common pharmacophores for a set of active molecules (optionally with
a volume union map).
The pharmacophore population profile normalised for the series
A table of molecules (rows) and pharmacophore populations (columns) for statistical analysis.
Options to reduce the size of the output are described in section 6.4.
The configuration bond rotations dialog is used
to set the rotational increments about the bonds.
The configuration advanced dialog is used
to specify the conformer generation mode and when this is not systematic (ie regular or random sampling)
the number of conformers required can be specified.
A pharmacophore is a 3D representation of the features in a molecule. It provides
a mechanism by which the important features of the molecule (ie those used in
drug-receptor interactions) can be represented without the "clutter"
of the other atoms in the molecule. This makes it easier to compare the features
across a set of molecules. The term centres is often used to refer to the
important features, and will be used in this chapter.
THINK uses 3-point (triangular) and 4-point (tetrahedral) pharmacophores. If
a molecule contains more than 3 or 4 important features then a set of pharmacophores
is obtained by taking all possible groups of 3 or 4 features from the molecule.
The distances between the points in the pharmacophore are calculated exactly,
but then allocated to distance bins. This allows the infinite range of
distances available to be represented by a finite set of values: each distance
is represented by the bin into whose range it falls.
THINK can also generate 2-centre pharmacophores. However, these
are only supported within site searches (see Chapter 7),
so are not described in this section.
6.1 Input Molecules
The input table is a set of molecules with a SDF column with 2D or 3D coordinates or a SMILES column.
THINK will automatically
generate 3D coordinates if they are absent from the input table. It is advisable to convert salts to
their parent forms and normal practice to eliminate molecules which are not drug-like using the Parent node.
The molecules must be read into THINK before their pharmacophores can be generated.
They may be read from any type of file. However, the user should be extremely
cautious about generating pharmacophores for a protein or peptide, since these
molecules are very flexible and can contain large numbers of centres, and therefore
may generate very large numbers of pharmacophores.
If a subset of molecules from a file is required, these may either be read
selectively from the file, or the whole file may be read and the desired molecules
specified as part of the pharmacophore calculation.
Commands
Dialogs
OPEN FILE=capsaicin.smi
File > Open
OPEN FILE=m2.sdf MOLECULE=TC1*
Selective read not supported
6.2 Set Pharmacophore Options
Most users favour 4-point pharmacophores, except for series of molecules where some
molecules have only a few centres when 3-point pharmacophores are more appropriate.
The implementation uses fuzzy pharmacophores which are designed to handle the tolerances on the exact positions of the centres (atoms) that define a pharmacophore, which affects the distances between the atoms.
When calculating a pharmacophore, THINK allows a tolerance of ±x on each distance (see THINK theory manual for more information).
Most users favour 4-point pharmacophores, except for series of molecules where some
molecules have only a few centres when 3-point pharmacophores are more appropriate. There is also an option to control whether THINK requires all hydrogen donors
to have an attached hydrogen atom before they are treated as centres.
The implementation uses fuzzy pharmacophores which are designed to handle the tolerances on the exact positions of the centres (atoms) that define a pharmacophore, which affects the distances between the atoms.
When calculating a pharmacophore, THINK allows a tolerance of ±x on each distance (see THINK theory manual for more information).
The definition of a set of pharmacophores consists of three main items:
The number of points (centres) in a pharmacophore (3 or 4)
The distance bins used
The feature types used
All these options can be altered through the CUSTOMISE command.
Most users favour 4-point pharmacophores, except for series of molecules where some
molecules have only a few centres when 3-point pharmacophores are more appropriate. There is also an option to control whether THINK requires all hydrogen donors
to have an attached hydrogen atom before they are treated as centres.
The implementation uses fuzzy pharmacophores which are designed to handle the tolerances on the exact positions of the centres (atoms) that define a pharmacophore, which affects the distances between the atoms.
When calculating a pharmacophore, THINK allows a tolerance of ±x on each distance (see THINK theory manual for more information).
The distance bins are used to transform the distances within each pharmacophore
(3 distances in a 3-point pharmacophore, 6 in a 4-point pharmacophore) into a
set of integers that give a more compact representation of the pharmacophore.
The upper limit of each distance bin should be specified (in ascending order)
with the BINS keyword, separated by commas with no intervening spaces ie BINS=a,b,c,d,e
where a,b,c,d,e are the bin limits.
By default, THINK will recognise all 12 supported feature types (HDON, HACC,
POS, NEG, AROM, LIP, ACID, BASE, MET, LPD, USR1, and USR4) in the molecule. THINK
can be tailored to ignore one or more of these atom types by specifying the list
of acceptable types with the TYPES keyword. The types should be supplied as a
list of 1-letter codes (D, A, P, etc) or centre names (HDON, HACC, POS, etc) separated
by commas and with no intervening spaces (eg TYPES=HDON,HACC,AROM,LIP).
In most circumstances the Boltzman population pharmacophore profiles are preferred which weight
each pharmacophore count by the energy dependent Boltzman population and express the result as a percentage.
There are options to use a simple count per conformer, a count per molecule and a percentage population.
Commands
Dialogs
CUSTOMISE CENTRES=3 ...
File > Preferences > Pharmacophores
CUSTOMISE DONOR=NOHYDROGEN COUNT=FRACTION ...
File > Preferences > Pharmacophores
Commands
Dialogs
CUSTOMISE CENTRES=3 ...
File > Preferences > Pharmacophores
CUSTOMISE BINS=3,4.5,6,7.5,9,12,15,18,21 ...
Not available
CUSTOMISE TYPES=D,A,N,P,R,L ...
Not available
CUSTOMISE DONOR=NOHYDROGEN COUNT=FRACTION ...
File > Preferences > Pharmacophores
6.3 Set Conformer Options
The pharmacophore calculation generates conformers of the molecules being processed,
and therefore is affected by the settings in effect for the conformer generation.
The user may wish to change the conformer search mode (systematic, sample, etc)
or to alter the number of increments sampled about each type of bond (single,
conjugated, ring, etc). See Chapter 5 for more details
on these settings.
The conformer contacts check is normally applied during pharmacophore calculations.
This can speed up the calculations by skipping all conformers
that contain CPK or VdW contacts. This does not affect the pharmacophore count
since only pharmacophores in accepted conformers are included.
The output from the pharmacophore node consists of one of the following:
Rows of 3D coordinates for each pharmacophores (SD format); pharmacophore identifier and population for use
by the Search node;
Pharmacophore profile consisting of the pharmacophore identifiers and their populations for use by the
FocusedSet and DiverseSet nodes;
A table of molecules as rows and pharmacophore populations as columns for use with general statistic nodes.
There are several options to reduce the size of the output table.
The number of pharmacophores output with 3D coordinates can be usefully limited to the most common 'n' pharmacophores.
In most cases, pharmacophores with low populations are of little interest and can be safely omitted.
The average population for a pharmacophore in a series of molecules is not always a reliable indicator that all molecules exhibit a pharmacophore because
the population for individual molecules may vary. Consequently, when creating maps for pharmacophores intended
to represent the union of volume occupied by all the active molecules when exhibiting that pharmacophore,
it can be useful to validate that most of the series of active molecules exhibit the pharmacophore.
The pharmacophores are automatically calculated when the user creates a pharmacophore
file or (less commonly) lists them to the console window. If the user wishes to
list or save the pharmacophores for selected molecules then their names should
be specified as part of the SAVE or LIST command.
The pharmacophore file is written using a compressed format that is described
in the THINK Theory Manual.
Commands
Dialogs
SAVE FILE=asp.phm
File > Save
SAVE FILE=M2.PHM MOLECULE=TC*
Selective calculation and save not supported
LIST INFO=PHARMS MOLECULE=TYR
View > Atom Data > Data:Pharms; Filter=TYR; List
If the molecules have not been previously filtered by drug-like or other desirable properties, this
can be performed by adding OPTIONS=FILTER to the SAVE command. Elimination of salts (ie
conversion to parent form) must be performed as a prior step.
Usage Mode:
7 DiverseSets and FocusedSets
This functionality is not implemented for Windows dialogs or command mode.
The DiverseSet node selects a subset of molecules which add to the pharmacophore diversity of
the starting pharmacophore profile.
The output is the table of selected molecules with SDF and SMILES columns.
The configuration options dialog
controls the diversity, pharmacophore centres and distance tolerances.
The configuration bond rotations dialog is used
to set the rotational increments about the bonds.
The configuration advanced dialog is used
to specify the conformer generation mode and when this is not systematic (ie regular or random sampling)
the number of conformers required can be specified.
The FocusedSet node selects a subset of molecules which exhibit pharmacophores
which overlap with those of the starting pharmacophore profile.
The output is the table of selected molecules with SDF and SMILES columns.
The configuration options dialog
controls the overap, pharmacophore centres and distance tolerances.
The configuration bond rotations dialog is used
to set the rotational increments about the bonds.
The configuration advanced dialog is used
to specify the conformer generation mode and when this is not systematic (ie regular or random sampling)
the number of conformers required can be specified.
7.1 Molecule and Pharmacophore Inputs
The input table of molecules must contain a SDF or SMILES column. If 3D coordinates are
not provided they will be generated automatically by THINK.
The implementation uses fuzzy pharmacophores which are designed to handle the tolerances on the exact positions of the centres (atoms) that define a pharmacophore, which affects the distances between the atoms.
When calculating a pharmacophore, THINK allows a tolerance of ±x on each distance (see THINK theory manual for more information).
The pharmacophore profile is an output of the Pharmacophore node. This should have the same
number of centres and distance tolerance.
7.2 Pharmacophore Options
The essential differences between the DiverseSet and FocusedSet nodes, is the criterion for
selecting molecules. To be included in the DiverseSet output, a molecule must exhibit a minimum
number of additional pharmacophores whereas in the FocusedSet node, molecules are selected if they
exhibit more pharmacophores which are in the starting profile than a default threshold.
Using 4-centre pharmacophores gives a larger number of possible pharmacophores and therefore
a larger measure of diversity. However, some molecules with little functionality may have too few
centres to exhibit 4-centre pharmacophores. The distance tolerance
The bond rotation and conformational generation options would normally be set the same as in
the Pharmacophore node for generating the starting profile.
7.3 Subset Calculation
Molecule selection based on pharmacophores tends to be slower than Searching or Docking because
all conformations have to be processed. As a consequence DiverseSet and FocusedSet nodes are
normally not used for hundreds of thousands of molecules. Progress reports are provided in the normal
way and the priority of the THINK process doing the calculation is lowered to ensure that the PC
remains responsive.
The configuration advanced dialog is used
to specify the conformer generation mode and when this is not systematic (ie regular or random sampling)
the number of conformers required can be specified. There is also an option to retain all conformers which
match a 3D or pharmacophore query rather than just the best.
Click on the box to display the corresponding dialog.
THINK can currently perform the following types of searches:
Search type
Description
Dialogs
2D Substructure
Checks connectivity for substructure within molecule
2D Exact
Checks connectivity for exactly the same molecule
R-group
A form of 2D search to find suitable reagents for a combinatorial chemistry
library. See chapter 15
Similarity
Compares the functional group keys and/or field data.
3D
Checks connectivity and centre-centre distances (requires the 3D module)
Pharmacophore
Checks centre-centre distances (requires the pharmacophore and 3D modules) and optionally a map volume constraint.
Site
A form of 3D search to find ligands that dock into a receptor site (requires
the pharmacophore and 3D modules). See chapter 9
An exact match 2D search checks the connectivity, bond order and elements (optionally
atom types) to determine whether the query molecule is the same as the molecules within
the file being searched. The substructure search differs by allow additional connections
to the query in the molecules being searched
3D and Pharmacophore searches perform the same connectivity checks as a 2D search matching all
the atoms in the query, and then
generates conformers of the target molecule (the molecule being compared with
the query) and compares the centre-centre distances in the query molecule with
the corresponding distances in the target conformer. The distinction between these searches
is only that a 3D search may contain more substructure and atoms which are not pharmacophore centres.
8.1 Query Molecule Input
The input table contains the query or queries in SMILES or SD format. Where multiple rows
are present only the first row is processed by default. However, occasionally for substructure
searches it can be useful to process multiple queries.
When a substructure or 3D search is to be performed, THINK will not add hydrogens to complete
missing valencies. For a pharmacophore search, the input is normally a selected row from the
output of the Pharmacophore node.
A second optional input is available for the table with a SDF or SMILES column of
molecules to be searched. For relatively small numbers of molecules
this offers an alternative to reading them from a file.
The molecule to be used as the query during the search is read from a SMILES,
PDB or SD file. It need not be the only molecule in the file - the user may either
read the entire file and then select the desired molecule.
If a substructure or 3D search is to be performed, it is recommended that the NOHYDROGENS
option is used to prevent THINK automatically adding hydrogens or interpreting
the hydrogen-count field in SD files to complete the molecule.
Commands
Dialogs
OPEN FILE=CAPQUERY.SMI OPTIONS=NOHYDROGENS
File Explorer Option
For 3D searches, the relative positions of the atoms within the query molecule are important
and consequently these must be read from a PDB or SD file since these contain 3D coordinates.
THINK does not support the MDL query file format.
8.2 Search Options
The molecules in an SD or SMILES format file may be searched directly without either reading them into
a KNIME table or a third party database by selecting the file to be searched. If the molecules to
be searched already exist in a KNIME table with a SDF or SMILES column, then this can be used for
the second (optional) input.
The filter option reduces the number of molecules examined during a search by eliminating
those that are not "drug-like" because containing undesirable substructures or property values.
Unlike many other software packages that require molecules to be converted
to a proprietary format before they can be searched, THINK searches molecules
directly from a SMILES or SD file. This eliminates the need to perform time-consuming
data conversions.
Commands
Dialogs
SEARCH ... FILE=capsaicin.smi ...
Search > 2D or 3D or Similarity > Browse
In command mode, the MODE keyword determines the type of search while the GUI provides
several alternative dialogs.
In all types of 2D and 3D searches, the
user has the option of matching atoms according to their element symbol or atom
type, with atom type matching being the more restrictive search. In addition,
the user can choose whether bond orders should be checked during the search.
The filter option reduces the number of molecules examined during a search by eliminating
those that are not "drug-like" or containing undesirable substructures or property values. This is done by
applying the filters contained in a learn file generated during an earlier
data analysis (see Chapter 10). The name of the learn
file is taken from the name of the field containing activity data (set via the
CUSTOMISE command); if the field is not set then the file "default.lrn"
in the THINK_EXEC directory will be used.
For 3D and Pharmacophore searches, the molecules being searched should be in their
neutral form rather than salt form without counterions present. Unless the file being
searched has been pre-processed, the Parent option should be enabled.
The settings used in the conformational
analysis (see Chapter 5) and the tolerance allowed
when comparing distances in the query with distances in the molecule are not used for
2D exact or substructure searches.
A table of hits (molecules which match the search criteria) with the SD and SMILES columns.
Summary information including the number of hits for each query.
Information about the failures including a brief summary of why they didn't match the query
The results of a search are stored in an output file that can have one of the
following formats:
SMILES - usually used for 2D searches
SDfile - used for 3D and site search results
Listing - a simple list of the names of the molecules that match the query
There are no restrictions on the format that may be used with each type of
search. The format of the input file (the file being searched) does not affect
the format of the results file (eg the results may be stored in an SD file even
though input file uses the SMILES format). The file format is deduced automatically
from the file name.
Commands
Dialogs
SEARCH ... OUTPUT=dopamine-s1.smi ...
Search > 2D or 3D or Similarity > Search
8.4 Perform Search
The time taken by a search is dependent on the numbers of molecules being searched and for 3D/Pharmacophore searches
the flexibility of these molecules. During a search the priority of the THINK process is lowered so that
other activities on the computer may be more responsive. The progress of the search is reported in the usual way.
When using the GUI, to perform an Exact match search the Exact option needs to be selected
on the 2D search dialog. Similarly on the 3D search dialog the Site option needs to be
selected to perform a Site search.
If insufficient information is specified for the search, the input file
does not exist or the output file
already exists etc, then an error message
is generated. For all types of searches, each molecule is read from
the input file in turn and compared with the query. If necessary, the molecule
will be converted to 3D before being compared.
For a 2D or normal 3D search, all the atoms in the query must be matched within
the molecule for it to be accepted as a hit and saved in the results file. In
a site search, only the atoms within one pharmacophore must be matched. This means
that several hits may be accepted from a single conformer of a ligand, if it can
be matched with more than one of the query pharmacophores.
The output tables can be viewed or analysed and the structures visualised by a range of KNIME nodes.
The THINK display node is designed for visualising 3D hits with a pharmacophore query together with
an optional volume map. It can also be used to visualise other search results in 3D or 2D.
If the search results are stored in a SMILES or SD file, they can be viewed,
listed and analysed in exactly the same way as any other set of molecules. The
query may be superimposed as the molecules are displayed, and the mapping between
the atoms in the query and the corresponding atoms in each hit molecule can be
shown using dashed lines. This requires both the query molecule and the hits to
be present in THINK (the query is not stored as part of the results file). In
THINK v1.25 the mapping can only be shown on 3D pictures.
View > Molecule > 3D Stick; Query:1IR3; Lines Show: Interactions
Usage Mode:
9 Maps
When using THINK from KNIME, the map calculation and display are integral parts of the
Display and Pharmacophore nodes when a volume constraint can be associated with each
pharmacophore. These maps can also be used as site contraints for Docking and De Novo derivative
generation when the pharmacophore is used as the search query. There is no separate Maps Node or KNIME
data type to support maps.
The most common use of the map functionality is to view a set of molecules with a map which
represents the volume occupied by one active molecule or the union volume of several active molecules which is
also known as a ligand volume map. Originally, this approach was used to
identify volume which was occupied by inactive molecules and not active molecules - this would be visible as atoms
outside the volume map. Inactivity can then be explained by asserting
that this space was preferrentially occupied by protein receptor atoms and therefore not available to ligands.
The use of Ligand Volume Maps is potentially exciting because they can give very reliable predictions. Unfortunately, the information
required for creating these maps is often only available when projects have progressed well beyond early stage discovery.
THINK provides additional capabilities including the use of a volume map to constraint the
results of a SITE search or a de novo derivative generation.
9.1 Read Molecules
The molecules are normally read from an SD file which were the results of a SITE search or less commonly a
3D search or a 3D de novo derivative generation. In these cases the molecules are already
aligned and the alignment step is skipped.
9.2 Align Molecules
The molecule alignment is the most critical step of the procedure and is dependent on the selection of
conformers and identifying features to overlap. It can be tempting and erroneous to align molecules based
on the reactivity of their functional groups rather than the potential interactions with a protein receptor.
The pharmacophore technology used in THINK is designed for pragmatically aligning molecules see Ligand Query Maps 9.5. For molecules
with little or no flexibility it is conceivable to generate appropriate conformations interactively
and align the molecules as described in section 4.2.
9.3 Calculate Map
A volume map may be constructed for a single molecule such as the most active or a Ligand map for series of molecules such
as those that exhibit submicromolar activity.
When selecting a Ligand map for active molecules it is necessary to configure the data field to be used,
the type of activity and the partitioning into actives and inactives. The Activity tab on the Preferences
dialog or the CUSTOMISE command provides these options. When partitioning molecules into actives and inactives,
either absolute activity measures can be used or a factor (0.1-1.0) can be specified when the activity range is
divided into 3 regions: most active; intermediate
and least active. Thus 0.4 would class the top 40% by value as active and the lowest 40% as inactive leaving the
mid range 20% as indeterminant.
It is sometimes apparent that molecules use different binding modes in the protein receptor site.
Under these circumstances some active molecules might within the volume map of the most active molecule
while others may not. This may be further complicated by molecules which can bind in more than one
mode - a default SITE search would only save one conformer per molecule. It may be possible to conclude that a binding mode is associated with activity
by comparing how a series of molecules bind and their activities. It is also possible to adjust the
SITE query so that results for only one binding mode are saved.
9.5 Ligand Query Maps
The Ligand Query procedure considers multiple overlap modes for a series of molecules and identifies
the pharmacophore and ligand volume map which best accomodates the set of active molecules. The approach
does not require knowledge of the protein receptor site although it relies on the active molecules
binding in precisely the same way. This can be validated by searching training sets and subsequently used to identify molecules which should exhibit
activity.
Usage Mode:
10 Docking
The identification of potential binding sites and the docking of molecules into a binding site are
two distinct steps in THINK provided by the FindSites and Docking nodes respectively. THINK uses
pharmacophore technology for docking which means that docked molecules will have well defined
interactions within the binding site. For the best results, the conformations of both the small molecule and the protein
are refined to give the best interaction score. One might expect a correlation between score, binding free energy and inhibition but
because of various assumptions and approximations quantitative predictions are not normally reliable. Typically, up to 10-30% of the molecules with the best scores exhibit µMolar activity.
The FindSites node reads in a PDB file and creates site queries based on docked ligands,
PDB binding site records and/or scanning the protein to locate binding pockets.
The output consists of
The Site Queries for use with the Docking node.
The Interactions with bound ligands which can be used as residue constraints in the Advanced Options.
A table of bound ligands which can be used to validate Site Queries with the Docking node.
The configuration Options Dialog
is used to specify the PDB file and select which creation modes are used etc.
The configuration Advanced Dialog
is used to specify residues with which interactions are required.
:
The Docking node attempts to dock molecules into the binding pocket defined by the site query.
The output is described below.
The configuration Options Dialog
is used to set the main docking options.
The configuration Bond Rotation Dialog
is used to change the bond rotational increments.
The configuration Advanced Dialog
is used when some more advanced options need are changed.
:
The promiscuity node performs multiple searches (normally in parallel) using a set of queries stored concatenated in a single
PDB format file. Normally the set of site queries are associated with selectivity or potential promiscuity of the ligands. Each site query
can have different configuration options such as the number of centres, warp speed etc which are stored in the file. See find-a-drug.pdb
for example.
Docking small molecules into protein binding sites and scoring how well the molecule interacts might indicate the potential of that
molecule to inhibit. (Note: Binding and inhibition are not necessarily the same). THINK includes capabilities to create site queries
for binding pockets, generate conformations for molecules and dock them using pharmacophore technology to the binding target where
the conformations of both small molecule and protein can be refined to give an optimum score.
The site query consists of a set of complementary centres such as hydrogen bond donors, acceptors,
charged atoms, ring centres etc that represent the types and positions of centres that may be present in a ligand if
it interacts in the protein receptor site. When there is a crystal structure with a known ligand this can be used to identify the site and
create the query. THINK also has functionality to find potential binding or active sites.
10.1 Creating Site Queries
A crystal structure of ligand-protein complex provides valuable information about one binding mode.
However, THINK's capability to search for all sites might identify additional residues in the same binding
pocket as the bound ligand which might interact with other ligands. In some cases, the PDB site records
are of little use, for instance when they define catalytic residues rather than a binding site.
When the precision of the crystal structure is low or the flexibility is high it can be adviseable
to increase the distance tolerance although this will inevitably give more hits in subsequent searches.
Residue constraints reduce the number of hits and without reducing the number of relevant hits if
there is also evidence that interaction with specific residues is associated with desirable biological
activity. In most cases, it is appropriate to review the Interaction table output from this node and view the binding
site using the Display node in order to be satisfied that requiring interactions with specific residues is appropriate.
As the Docking node has no functionality to select a site query from Sites output table, it is common to
use a generic KNIME node to filter this output.
The part of the protein or binding site in which molecules are docked is identified by:
Using a known bound ligand
The SITE record in a PDB file
THINK functionality to scan a protein and identify binding pockets.
A default query for a site search may be
created within THINK and stored as a PDB file but it may be necessary to edit this
file to shift the coordinates of centres, remove centres, indicate required centres, adjust tolerances
etc. Further information can be found in the THINK theory manual.
The query and the active site of the protein can be saved to the same file.
Commands
Dialogs
MODIFY INTERACT=^1B7Y LIGAND=PAA5P SAVE FILE=1B7Y-Q1.PDB MOLECULES=INTERACT,1B7Y
Edit > Site > Ligand:PAA5P; Create; Save
The extended active site is often
used to provide supplementary data to the query, for instance to ensure that ligands
found during the search do not occupy the same space as the protein itself. The
protein, or the subset of the protein around the receptor site, should be read
before the search is initiated.
10.2 Docking Inputs
The Site query created by the FindSites node is a required input which defines both the interactions
and the residues in the binding site. If the set of molecules being searched for potential ligands is
relatively small these can be supplied as a table with a SDF or SMILES column as the second input. Otherwise
a potential ligand file may be configured in the Options dialog.
The site query is read from a PDB file created by the command sequence describe earlier. The file of
potential ligands to be searched is specified on the Search Dialog or using the FILE keyword of the
SEARCH command.
10.3 Docking Options
The molecules being searched should be in their neutral form rather than salt form and counterions
should be absent. The Parent option should be enabled unless the molecules are known to be
in the correct form. The scoring function considers permutation of possible charge and tautomeric
states with the consequence that this does not require explicit thought or action by users! In many
cases, it is appropriate to apply drug-like filters which will reduce the number of inappropriate hits.
The pharmacophore docking algorithm will often give more hits, the lower the number of
centres in the pharmacophore (see Chapter 6) and the larger the distance tolerance with increasing elapse time
unless residue constraints are being used.
The quality of the hits in terms of the proportion of true positives, is often significantly
increased by permitting torsion refinements of the torsion angles in the ligand and the
protein side-chains. However, accomodating such flexibility tends to increase the elapse time
significantly.
The warp speed control reduces the search time at higher warp values by skipping some potential
hits which interact with residues which are more distant from the original ligand used to create
the query or the centre of the binding site (in the absence of a bound ligand). Details of this
control algorithm are found in the THINK theory manual. The search
speed can often be improved without loss of classes of hits by prohibiting more than one interactions
with each protein ligand.
Several of the more advanced controls for docking, especially those that effect pharmacophores in
general, use the CUSTOM command or the PREFERENCES dialog, The 3D search dialog with the SITE option
is used to perform docking.
The Docking includes a conformational search step for the ligands and the bond rotations
and related conformational generation options can be controlled in the usual way (see Chapter 5).
There are also time limit and option to retain multiple hits in different conformers for each
molecule (rather than the conformer with the best score). The volume (4-centre) and area (3-centre) minimum
fractions reject molecules which exhibit the pharmacophore using only a small part of the ligand.
THINK will automatically calculate a score for each hit using an
extended ChemScore function which is stored
the field G-TOT and the contributions in other fields. Although this value is only an approximate indication of
the free energy of binding, more negative values suggest stronger binding.
After geometry refinement, any hit whose score exceeds a threshold is rejected.
Commands
Dialogs
CUSTOMISE CENTRE=3 SEARCH MODE=3D CUTOFF=-30 ...
Search > 3D
10.4 Search Results
The output consists of 3 tables:
A table of hits (molecules which match the search criteria) with the SD and SMILES columns.
Summary information including the number of hits for each query.
Information about the failures including a brief summary of why they didn't match the query
The results of a search are normally stored in a 3D SDF output file. This uses serial numbers to map
the atoms to the corresponding atoms in the site query and the protein. The conformational numbers ae used to
distinguish different conformers without implying a separation in conformational space.
10.5 View Docking Results
The output tables can be viewed or analysed and the structures visualised by a range of KNIME nodes.
The THINK display node is designed for visualising 3D hits with or without the site query and
binding site of the protein. The query and/or the protein may be superimposed as the molecules are displayed, and the mapping between
the atoms in the site query or the protein and the corresponding atoms in each hit molecule can be shown using dashed lines.
If the search results are stored in a SMILES or SD file, they can be viewed,
listed and analysed in exactly the same way as any other set of molecules. The
query may be superimposed as the molecules are displayed, and the mapping between
the atoms in the query and the corresponding atoms in each hit molecule can be
shown using dashed lines. This requires both the query molecule and the hits to
be present in THINK (the query is not stored as part of the results file). In
THINK the mapping can only be shown on 3D pictures.
View > Molecule > 3D Stick; Query:1IR3; Lines Show: Interactions
Usage Mode:
11 Property Diversity
The THINK functionality to visualise property diversity and to select subsets based on property
values is not accessible from KNIME.
The property diversity examines the volume, lipophilicity and number of centres
in a set of molecules and determines the diversity of the set from these factors.
There are two routes by which the diversity of a set of molecules may be calculated.
One route takes molecules directly from a file, and allows the user to save the
set of selected (diverse) molecules in a results file. This route provides more
flexibility in changing the calculation options. The other route uses molecules
currently loaded within THINK and is designed to show the diversity of the current
molecules, rather than to select a diverse subset.
In the following sections, the number in parentheses in each title indicates
the route to which the section refers.
11.1 Identify File to be Processed (1)
In route 1, the set of molecules to be analysed is read directly from a SMILES
or SD file - there is no need for the molecules to be read into THINK before the
calculation is performed. The file format is deduced automatically from the file
extension supplied as part of the file name. The molecules could be read from
a PDB file, but this format is normally used only for peptides and proteins, which
are not suitable for property diversity calculations.
Commands
Dialogs
SELECT FILE=capsacin.smi ...
Calculate > Selection > File or Browse
11.2 Read Molecules to be Processed (2)
If route 2 is being used to calculate diversity, the molecules must be read
into THINK before the calculation is initiated. If a subset of molecules from
a file is required, these may either be read selectively from the file, or the
whole file may be read and the desired molecules specified as part of the diversity
calculation.
Commands
Dialogs
OPEN FILE=capsacin.smi
File > Open
OPEN FILE=m2.sdf MOLECULE=TC1*
Selective read not supported
11.3 Set Calculation Options (1)
The diversity calculation uses a three-dimensional property space plotting
molecular volume, lipophilicity and the number of centres (0-24) along the three
axes. Each axis is divided into 25 cells; thus using 253 cells to cover the entire
property space. During the calculation each molecule is allocated to a cell unless
that cell is already fully occupied, in which case the molecule is rejected. The
user can limit the maximum number of molecules allowed in a cell.
Normally, the diversity calculation would start from an empty property space.
However, the user may choose to append the current calculation to the results
of a previous calculation, in which case the cell occupancies from the previous
diversity calculation are retained and used as the initial values for the current
calculation.
Commands
Dialogs
SELECT ... LIMIT=3 ...
Calculate > Selection > Limit=3
SELECT ... MODE=ADD LIMIT=5 ...
All options not available
11.4 Supply Results File (1)
A simple list of the molecules selected by the diversity calculation can be
stored in a results file. Alternatively, the selected molecules themselves may
be stored in a SMILES or SD file. The file format required is deduced automatically
from the file name.
Commands
Dialogs
SELECT ... OUTPUT=m3div.set
Calculate > Selection > Select
11.5 Select Plot Style (1,2)
The diversity of the selected molecules can be shown in a panel plot or a 3D
cube plot. In the former, the plot is divided into 25 separate tiles or individual
plots, each representing a different number of centres. Within each tile, volume
is plotted along one axis and lipophilicity along the other.
In a 3D cube plot, volume, lipophilicity and the number of centres are plotted
along the three axes of a cube that can be rotated.
Commands
Dialogs
SELECT ... DISPLAY=PANEL ...
Calculate > Selection > Panel
SELECT ... DISPLAY=CUBE ...
Calculate > Selection > Cube
DISPLAY ... MODE=PANEL ...
View > Diversity > Panel
DISPLAY ... MODE=CUBE ...
View > Diversity > Cube
11.6 Define Activity-Based Colour-Coding (2)
When route 2 is used, the user may opt to colour-code the molecules in the
diversity plot according to their activity. This can only be achieved if activity
data was loaded with the molecules. The user must supply the name of the field
containing the data.
Commands
Dialogs
DISPLAY ... ACTIVITY=EC50
View > Diversity > Field:
11.7 Calculate Diversity (1,2)
In a route 1 diversity calculation, each molecule is read in turn from the
file. The volume, lipophilicity and number of centres are calculated, and the
molecule is then allocated to a cell in the property space. If the cell is already
full the molecule is rejected, otherwise it is accepted and added to the list
of selected molecules. Thus, the order of the molecules in the input file may
affect the final selection.
The method used in a route 2 calculation is similar, except that the molecules
do not need to be read from an input file. There is no cell limit in a route 2
calculation, so all molecules are retained and placed in the diversity plot. At
the end of the calculation the molecule represented by any point on the plot may
be viewed by picking that point.
The THINK functionality for Data Analysis is not accessible from KNIME.
The data analysis examines the properties and/or keys of a set of molecules
and attempts to identify features that may be responsible for undesirable characteristics.
The results of the analysis are stored in a learn file that can be used
in subsequent THINK calculations to highlight or eliminate molecules whose properties
lie outside the acceptable ranges or which contain unacceptable functional groups.
12.1 Read Molecules
THINK will analyse all molecules that have been loaded into the program. They
may be read from a SMILES or SD file. Any external property data required for
the analysis must be included in the file in data fields - these fields will be
loaded automatically as the molecules are read. The file must contain a field
of activity data.
Commands
Dialogs
OPEN FILE=capsaicin.smi
File > Open
12.2 Select Activity Field
The field containing the activity data must be identified.
Commands
Dialogs
LEARN ... ACTIVITY=EC50 ...
Calculate > Analysis > Field:EC50
12.3 Set Activity Options
By default, the activity values will be taken directly from the activity field,
with the highest values indicating the most active molecules. Alternatively, the
interpretation of activity may be reversed, so that molecules with low values
are considered the most active. There is also an option to convert the activities
into their logarithmic values, for instance if binding constants are used as the
activity data.
The molecules need to be divided into active and inactive molecules for the
analysis. This is done through a user-supplied significance value that must lie
within the range 0-0.5. Molecules that lie within this fraction of the top or
bottom of the activity range are considered active or inactive; molecules that
lie in the middle are ignored during the analysis. If a significance value of
0.5 is specified then all molecules will be included in the calculation.
Commands
Dialogs
LEARN ... SIGNIFICANCE=0.3 ...
Calculate > Analysis > Significance=0.3
LEARN ... OPTIONS=LOW,LOG ...
Calculate > Analysis
12.4 Select Data to be Analysed
THINK can include some or all of the functional group keys, 2D properties and/or
external data fields in the data analysis. The fields, 2D properties and keys
are identified by keywords:
Keyword
Interpretation
FIELDS
All 2D properties and external data fields
field_name
Specified external data field
property_name
Specified 2D property. Valid names are:
ATOMS
BONDS
HETATOMS
HALOGENS
DONORS
ACCEPTORS
POSITIVES
NEGATIVES
BRANCHES
RINGS
AROMATICS
HETAROMATIC
CENTRES
MASS
FLEXIBILITY
LIPOPHILICITY
VOLUME
AREA
PSA
NPSA
PFA
NPFA
XSA
XFA
CPK-CONTACTS
VDW-CONTACTS
ROT-BONDS
CONFORMERS
E-TORSION
KEYS
All functional group keys
KEY#n
Specified functional group key
*
All functional group keys, 2D properties and external data fields
<no keywords supplied>
All functional group keys, 2D properties and external data fields
See section 4.4 for a full list of the 2D properties.
Commands
Dialogs
LEARN ... PROPERTIES=KEYS ...
Calculate > Analysis > Keys
LEARN ... PROPERTIES=KEY#5,KEY#12,LOGP,DIPOLE,...
Full control is not supported
12.5 Analyse Data
During the analysis calculation, THINK first calculates any 2D properties and
functional group keys that are required before attempting to extract discriminating
features that could identify the inactive molecules. The results of an analysis
calculation are saved in a learn file whose filename is taken from the name of
the activity field and uses the file extension ".lrn". Thus, if the
activities are taken from the field called EC50, the results will be stored in
a file called "ec50.lrn". Amongst other information, the learn file
contains the acceptable range of values for each discriminating property (2D property
or external data field) and the most significant unacceptable functional groups.
The learn file generated by data analysis may be used to provide additional
rejection criteria for de novo structure generation. It may also be used in the
property spreadsheet to highlight values that lie outside the acceptable ranges.
Commands
SUGGEST MOLECULE=M2 DISPLAY=PANEL ACTIVITY=EC50
LIST INFO=PROPERTIES OUTPUT=WINDOW ACTIVITY=EC50
Usage Mode:
13 Clustering
The Cluster node is useful in order to find representative molecules from a larger set such as
the results from a search. The functional group keys are used for the same similarity measure that
is used for similarity searching.
Click on the box to display the corresponding dialog.
A hierarchical clustering algorithm has been implemented in THINK which is appropriate to cluster
a few thousand molecules. This clustering capability is useful in order to find representative
molecules from a larger set such as the results of a search. The functional group
keys are used for the same similarity measure that is used for similarity searching.
13.1 Input Molecules
The input table is a set of molecules with a SDF column with 2D or 3D coordinates or a SMILES column.
THINK will analyse all molecules that have been loaded into the program. They
may be read from a SMILES or SD file.
Commands
Dialogs
OPEN FILE=capsaicin.smi
File > Open
13.2 Set Cluster Options
The clustering algorithm
works in two modes:
Fixed number of clusters (>0) or
Natural clusters which are defined by the similarity cutoff and requires the number of
clusters to be set to 0.
Commands
Dialogs
Output
CLUSTER SIZE=0 SIMILARITY=0.75
Calculate > Cluster > Numbers=0 Similarity=0.75
CLUSTER SIZE=20
Calculate > Cluster > Numbers=20
13.3 Cluster Molecules
The clustering algorithm starts with each molecule in a separate cluster and then
merges the two clusters which have the lowest maximum separation between any two molecules in
the clusters. This merging continues iteratively until either the desired number of
clusters have been formed or the lowest maximum separation is greater than the similarity cutoff.
In natural clusters, all the molecules in a cluster are at least as similar to each other
as the specified similarity. When the number of clusters is specified is larger than
the number of natural clusters, it is conceivable that molecules in a cluster have a low degree of similarity.
The output tables contain the molecules with columns for SD file, SMILES and the cluster identifier in a
column named ClusterID. The first table contains all the molecules while the second table only contains a
representative for each cluster.
A numerical identifier for each cluster is stored in a field named Cluster-ID and representive
for each cluster is placed in the Selected set of molecules.
13.4 View Clusters
This capability is not available from KNIME.
A dendogram is produced showing the hierarchy of clusters. The
molecule corresponding to the Y-coordinate on the dendogram is displayed by clicking. The up and down
arrow keys can then be used to display other molecules in that cluster.
If the spreadsheet or tile display is used, the selected molecules are the representives
of each cluster. It can sometimes be useful to sort the spreadsheet eg on Cluster-ID so
that the members of a cluster can be selected and saved.
This capability is not available in command mode.
Usage Mode:
14 De Novo Molecule Generation
The De Novo node generates derivatives of a starting molecule or molecule(s).
The Options dialog
is used to set the mode and control the number of molecules generated. In addition, this dialog allows
some fine control of the drug-like filters.
The output consists of a table of molecules with 2D or 3D SD column and a SMILES column. In addition
to various molecular properties, an index to the last transformation is included.
A genetic algorithm is used to generate the new molecules which in 2D mode uses a set of
transformations which can substitute groups, increase or decrease chain lengths etc. In 3D mode, the transformations
are limited to functional group substitutions via a single attachment atom. Drug-like substructure and
property filters
are always applied to the molecules generated.
Click on the box to display the corresponding dialog.
De novo molecule generation uses a genetic algorithm to generate derivatives
of a starting molecule using a set of user-defined rules to substitute functional
groups, increase or decrease chain lengths, etc. The new molecules are checked
for undesirable substructures or properties before passing though an annealing
process which constrains the generated molecules to be close derivatives.
14.1 Input Molecules
The input table is a set of molecules with a SDF column with 2D or 3D coordinates or a SMILES column.
Normally only the first molecule in the table is used.
De novo generation uses an existing molecule as the starting template.
This molecule, often referred to as a query, is usually read from a SMILES or
SD file. It need not be the only molecule in the file - the user may either read
the entire file and then select the desired molecule, or may selectively read
just the query molecule from the file (see section 2.1).
Commands
Dialogs
OPEN FILE=capsaicin.smi
File > Open
14.2 Set Molecule Creation Options
The initial molecule is converted into the potential new molecule by applying
a transformation chosen at random from the transforms file. A default file, "transform.smi",
is provided in the THINK_EXEC directory, but the user may supply a customised
SMILES file containing their own transformations. The format of the file is described
in the THINK Theory Manual.
Unless otherwise specified, de novo generation will continue until 1000
acceptable new molecules have been created.
The modifications may be restricted to specific atoms or groups of atoms by setting the group number for the atoms and then
restricting modifications to the atoms in that group.
The transformations are restricted in 3D to the subset that make substitutions at a single atom. With
map or protein constraints, the substitutions are further restricted to those which fit inside the volume
of the map or protein cavity after randomly sampling conformations.
If residue constraints are specified, then these are used to calculate a merit score for each derivative retaining the conformation
which achieved the best score. The merit is scaled in the range 0-100 for each residue based on distances between the atom pairs.
In the current implementation, the optimum distance between complementary centres such as those forming hydrogen bonds is 3Å which results
in a merit score of 100 which reduces on a linear scale to 0 at 8Å. For other atoms, the highest merit score 50 which corresponds to a
separation distance of 2Å (the contacts check eliminates lower separation distances). The merit score for each residue is the maximum of
that for any of the atom pairs and the overall merit for a conformer is determined by adding together the merit scores for each residue.
Thus merit scores of 300 or more are possible providing the numbers of residues in lists is sufficient for 3 or more interactions.
When starting from a 3-D docked small fragment, increasing the curb on non-H atom insertions allows molecules
to grow significantly and become drug-like. Small values of curb ensure that the derivatives have similar
numbers of non-H atoms to the starting molecule.
The parts of the molecule being modified can also be restricted by atom group number. These can be
set by the user with the Display Node although for 3D docked starting molecules (and other 3D hits) the
atoms not matched to the query are set to a group number of 999 and changes are automatically
restricted to the unmatched atoms.
The parts of the molecule being modified can also be restricted by atom group number. These can be
set by the user with the Display Node although for 3D docked starting molecules (and other 3D hits) the
atoms not matched to the query are set to a group number of 999.
14.3 Generate Molecules
De novo generation starts by taking the initial molecule and applying
to it a transformation selected at random from the transforms file to create a
potential new molecule. This molecule is then checked against the drug-like filters
before progressing to the annealing stage. This annealing step acts as a soft constraint
of the molecular properties to the starting molecule. The starting molecule
for the next loop of the molecule generator is randomly chosen from the resultant molecules and the
original molecule.
A diversity plot is displayed as the molecules are generated.
Usage Mode:
15 Combinatorial Chemistry
THINK uses a reaction based representation of a library.
This consists of a generic reaction with reactants including
substitution positions and product(s) which form the core group for enumeration. A SMILES
for creating an amide library is shown below together with a graphical representation.
[1]C(=O)Cl+[2]N(H)H>[1]C(=O)N(H)[2]
For most libraries there are several different generic reaction schemes which can have subtle
but important differences in the reagents which are selected. Probably the most innovative use of
this functionality in Fragment Based Drug Design (FBDD) to grow the initial molecule into a potential drug molecule.
The input for the Rgroup node is a reaction query. The reagents may either be read from
a file or supplied as a second (optional) input. A separate Rgroup node is used for each Rgroup in
the reaction query.
The Options dialog
dialog is important to select the R-group the node should process.
The output from the Rgroup node is a table of R-groups with generic connections intended for
connection to the appropriate input of the Enumeration node.
The first input for the Enumeration node is a reaction query. The number of other inputs which
need to be connected depends on the number of R-groups in the reaction query.
The Options dialog
dialog is important to select the number of R-groups the node should process.
The output from the Enumeration node is a table of molecules.
Combinatorial chemistry consists of two distinct steps:
Performing one or more R-group searches to convert reagents into R-groups
(requires the 2D module)
Enumerating the molecules by "plugging together" R-groups from two
or more files to form a library
R-groups are molecular fragments that can be plugged together to form larger
molecules. These joins can only be made at specific locations within the fragments:
the connection atoms. Each connection atom is a temporary atom with a special
numeric atom type in the range 0-9. When two R-groups are joined, the paired connection
atoms are removed and the remaining bonds are plugged together. The connections
may be made using any type of bond (single, double, etc), but both connection
atoms must have the same type of bond. If n is greater than 0 then that
connection can only be joined to another with the same value, and is known as
an explicit connection atom; a value of 0 indicates a generic connection
that can be joined to a connection atom with any value. A maximum
of 10 sets of R-groups, or 9 sets of R-groups and a set of core molecules, may
be used in any library.
R-groups may be stored using the SMILES or SD format. Within the SMILES string
each connection atom is shown as a single digit within "[]" brackets,
"[n]" where n is the numeric atom type. In an SD file,
each connection atom has an atom symbol of n, stored in columns 32-34 of
the atom record.
When a library is enumerated, THINK permutes all combinations of the R-groups
within the sets and saves the resulting molecules to a file.
More information about R-groups and enumerated libraries can be found in the THINK
Theory Manual.
15.1 R-group Search Input
The input table defines a reaction query with a SDF column with 2D coordinates or a SMILES column.
This can be created using the Display node or third party software such as the Marvin sketcher from
ChemAxon and its R-group designations. A second optional input is available for the table of
reagent molecules with a SDF or SMILES column. For relatively small numbers of reagents this offers an
alternative to reading them from a file.
It is also possible to use just the corresponding reactant of the generic reaction query as
the input instead, but in most cases this offers no advantages.
The molecule to be used as the query during the R-group search is read from
a SMILES or RD file. It need not be the only molecule in the file - the user may either
read the entire file and then select the desired molecule, or may selectively
read just the query molecule from the file (see section 2.1).
The SMILES string used to search the reagents is known as an R-group query and consists of a substructure and the connection
points. The substructure is the portion that is common to all reagents accepted
by the search; this portion will be discarded when the reagent is converted into
an R-group. The connection points are indicated by numeric atom types or wildcard
atom types:
[0] for a generic connection atom
[n] for an explicit connection atom
[wildcard] for a generic connection atom
The [0] and [n] connections in the query will be matched to any atom
type except hydrogen in the reagent. The [wildcard] connection will only
be matched to selected atom types - see the THINK
Theory Manual for a full list of supported wildcards. The [0] connection is not usually
used in reactions but is a useful means of generating R-groups which can be re-used with in a variety
of libraries.
It is also possible to store the R-group queries in separate molecules. For example,
to build a coreless amide library of the form [R1]C(=O)N[R2] from
acid chlorides and amines would require two R-group queries: one to look for
acid chlorides and convert them into suitable R-groups, and the other to perform
the same operation on amines. The reagents to be used, and the R-group query required
to select them are listed in the table below:
Search
Reagent
Query
acid chloride
[R1]C(Cl)=O
[0]C(Cl)=O
amine
N[R2]
[0]N
If the query contains multiple connection points, THINK will use explicit connection
atoms in the resulting R-groups, even though the the query may have contained
generic connection points. Since the order in which explicit connections are allocated
to generic connection points is undefined, it is recommended that explicit connections
([n]) are used in these queries.
Commands
Dialogs
OPEN FILE=rx-amide.smi
File > Open
Note: Reactions and R-group queries should never have missing hydrogens as these cause
serious problems.
15.2 R-group Search Options
It is necessary to specify which R-group is to be created by each Rgroup node.
The set of molecules for the R-group search can either be stored in a file, when its file name
needs to be configured or connected to the second optional input. This input must have a
SDF or SMILES column.
Many files of reagents store their molecules in the salt form and this will
cause spurious results from R-group searches. The THINK R-group
search incorporates the ability to convert the molecules into their parent forms.
This conversion takes place before the reagent
is compared with the query.
If the drug-like filter option is used, the upper limit is applied to the resulting R-group
(not to the original molecule), and the lower limit is ignored in order to avoid
eliminating solutions that could be satisfied in enumerated molecules by other
R-groups.
In addition to selecting the molecule which contains the R-group for reaction queries
it is necessary to specify which R-group to use. The SITE keyword is used to specify
any atom in the R-group (usually the connection atom by atom name ie without the square brackets).
Commands
Dialogs
SEARCH ... QUERY=rx-amide#1 SITE=2 ...
Search > 2D > R-group; Site=2
Unlike many other software packages that require molecules to be converted
to a proprietary format before they can be searched, THINK searches molecules
directly from a SMILES or SD file. This eliminates the need to perform time-consuming
data conversions.
Commands
Dialogs
SEARCH ... FILE=aldrich.smi ...
Search > 2D > Browse
An R-group search is a specialised form of 2D search (see Chapter 7)
that checks connectivity and automatically converts matching molecules into R-groups.
The user can choose whether atoms should
be matched according to their element symbol or atom type and bond orders
should normally be checked; these settings may be
controlled through the OPTIONS=ORDER and OPTIONS=TYPE keywords respectively.
Many files of reagents store their molecules in the salt form and this will
cause spurious results from R-group searches. The THINK R-group
search incorporates the ability to convert the molecules into their parent forms
through the OPTIONS=PARENT keyword. This conversion takes place before the reagent
is compared with the query.
Commands
Dialogs
SEARCH MODE=R-GROUP ... OPTIONS=ORDER,PARENT
Search > 2D > R-group; Order; Parent
If the FILTER option is used, the upper limit is applied to the resulting R-group
(not to the original molecule), and the lower limit is ignored in order to avoid
eliminating solutions that could be satisfied in enumerated molecules by other
R-groups.
15.3 R-group Search Output
The output consists of 3 tables:
A table of R-groups in the SD and SMILES columns.
Summary information including the number of hits for each query.
Information about the failures including a brief summary of why they didn't match the query.
The R-groups resulting from an R-group search are stored in an output file
using the SMILES or SD format, depending upon the file extension supplied for
the output file. Normally the SMILES format would be used because this is more
compact that the SD format.
Commands
Dialogs
SEARCH ... OUTPUT=gp2.smi ...
Search > 2D > Search
15.4 R-group Search
The time taken by a search is dependent on the numbers of molecules being searched
During a search the priority of the THINK process is lowered so that
other activities on the computer may be more responsive. The progress of the search is reported in the usual way.
Once the query molecule, reagent and R-group files have been supplied, the
user can initiate the search. Each reagent molecule is read from the file in turn
and compared with the query; if it matches then it is converted into an R-group
and saved to the R-group file. THINK automatically eliminates duplicate R-groups
- this situation would occur if the reagent file contained several copies of the
same molecule, for instance in different salt forms. THINK will also automatically
ignore all molecules with multiple reaction sites since it would be impossible
to predict which reaction site is used when physically making the library.
The enumeration inputs are the Reaction Query (where the core of the library is the product)
and at least one R-group input. The remaining R-group inputs are optional. It is important that
the number of connected R-groups match those needed for the library and that these R-group tables
are connected to the correct inputs. The R-groups generated by the Rgroup node are generic and
can be connected in any substituent position.
One some occasions it may be desirable to read R-groups from files in which the results of previous
R-group searches have been saved using the Open node and connect these to the Enumeration inputs.
For Fragment Based Drug Design, the simplest core in the bound fragment (with the 3D coordinates
to interact with the protein). Alternatively, a different core group can be used together with
an optional ligand (with 3D coordinates to interact with the protein) to which the core is fitted. In both
cases it is essential that the core is modified to indicate which atoms are the group connections for which
there is an optional dialog.
Once the R-groups for a desired library have been created, they can be used
to create the complete molecules. To achieve this, the R-groups must first be
read into THINK. The core group (if any) is treated as an R-group, and therefore
must also be read into THINK. The generic reaction (which contains the core group as a product)
may be used as the reactants will be ignored. Since there may be a large number of R-groups, it
is recommended that all other molecules are first deleted to reduce the amount
of memory required.
For Fragment Based Drug Design, the simplest core in the bound fragment (with the 3D coordinates
to interact with the protein). Alternatively, a different core group can be used together with
an optional query substructure (with 3D coordinates to interact with the protein but typically without hydrogens)
to which the core is fitted. In both cases it is essential that the core is modified to indicate which atoms are the
group connections for which the 2D Edit functionality or keyboard commands may be used.
The R-group files may be read in any order. THINK will use all the R-groups
loaded from each file to enumerate the library. If a subset of R-groups is required
from a file, then either the required R-groups should be read selectively (see
section 2.1), or the entire file should be read
and the undesired R-groups deleted.
Note that all the R-groups to be used at any single position in the library
must be read from the same file. This is because THINK uses the file name to identify
the R-groups for each position in the enumerated molecules. The R-groups from
a file may be used at more than one position in the library (eg when building
peptides), providing exactly the same set of R-groups is used in each position.
Commands
Dialogs
OPEN FILE=rx-amide.smi
OPEN FILE=g1.smi
File > Open
DELETE MOLECULE=PRO@G1
Edit > Delete
OPEN FILE=g2.smi MOLECULE=A*
Selective read not supported
Before a library can be enumerated, THINK needs to know which R-groups are
to be attached to each position. The R-groups are identified by the name of the
file from which they were read (when any molecule is read, THINK maintains a record
of the file from which it came). Only the file names are required (not the file
extensions).
If the R-groups contain generic connection points ([0]) then it is important
that the file names are supplied in the correct order, with the core file (if
any) first. If all the R-groups contain explicit connection points ([1], [2],
etc) then the files can be supplied in any order. If the same set of R-groups
are to be used at several positions, for instance when building a peptide, then
the file name must be supplied once for each position (as in the second example
below).
Commands
Dialogs
SAVE ... R-GROUPS=amide-rx,gp1,gp2
File > Enumerate
SAVE ... R-GROUPS=ncap,amino,amino,amino,amino,cap
15.6 Enumeration Options
It is essential to configure the Enumeration node so that the correct number of inputs is
processed by selected the number of R-groups in the library core.
An enumerated library may contain many thousands of molecules,
many of which are of no further interest because they have undesirable properties.
Many of these can be filtered out by using the Drug-Like filter option.
If the protein site is connected, then it will be used as a volume constraint for
the enumeration and the conformation of the enumerated molecule randomly sampled.
In addition, if any residue restraints are specified then a merit score (see section 14.2)
will be calculated and the conformation with the highest merit score will be retained.
An enumerated library may contain many thousands (or millions) of molecules,
many of which are of no further interest because they have undesirable properties.
These molecules may be filtered out from the library by applying the criteria
from a learn file. This file, created during an earlier data analysis calculation
(see Chapter 11), contains a list of undesirable substructures
and of desirable property values. When applied during library enumeration, molecules
which contain any of the undesirable substructures or whose properties lie outside
any of the desirable ranges are automatically discarded instead of being saved
to the file. Note that these filters are applied to the enumerated molecules,
not the constituent R-groups. The learn file is assumed to have the same name
as the field that contains activity data (set through the ACTIVITY keyword in
the CUSTOMISE command). If this field is not set then the learn file "default.lrn"
in the THINK_EXEC directory will be used.
Commands
Dialogs
CUSTOMISE ACTIVITY=LOGK
Not supported
SAVE ... OPTIONS=FILTER
File > Enumerate > Filter
If a query is specified, then it will be used to orientate the core group unless the reserved name
of CORE is specified, in which case the initial core orientation is preserved. Any protein will then be
used as a volume constraint for the enumeration and the conformation of the enumerated molecule randomly sampled.
In addition, if any residue restraints are specified then a merit score (see section 14.2)
will be calculated and the conformation with the highest merit score will be retained.