This release includes the ability to perform P450 profiling more information can be found in our white paper.
In addition, there are some enhancements and minor code changes:
The cavity map code allows the cavities used by the binding site search to be visualised (and in essence uses the same algorithms to deduce cavities). Functionality has also been added to allow maps with the same X,Y,Z extents and grid size to be combined logically and arithmetically. At present this functionality is restricted to command mode (ie not available from KNIME or native THINK dialogs).
| Commands | Dialogs | |
| CALCULATE MAP=CAV1 TYPE=CAVITY MOLECULE=1OG5 GRID=1 | Not supported |
There have been some changes to the ring fusion code reducing the number of instances when builds fail due to incorrect chirality (problems remain with some rings which are bridged and fused).
Inter and Intra contact colouring
The inter and intra contacts from the molecule designated as the query are now coloured differently.
Additional secondary structure colouring options
The Display Panel now includes additional options to colour by hydrophobicity, secondary structure type and charge.
Analogously to Volume for 3D queries (4 centre pharmacophores) and Area for 2D queries (3 centre pharmacophores), Extent has been implemented for 2 centre queries which will eliminate some potential hits were the pharmacophore is exhibited by a small subset of the molecule. As a result 2D searches are significantly faster.
There was an obscure bug which caused the File Explorer dialog to be unresponsive under Windows 8 (no problem under XP). This has now been worked around.
This release includes some significant enhancements for Fragment Based Drug Design (FBDD), examples of which can be found in white paper.
In addition, there are improvements to the performance 3D and Pharmacophore searching.
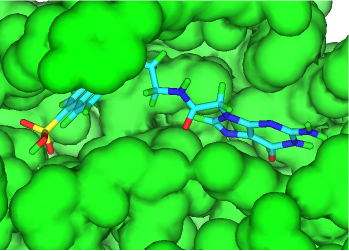
Key algorithms used by the 3D De Novo capabilities have been rewritten to improve performance and add extra functionality when small molecules or fragments docked into a protein binding site are grown into larger drug-like molecules. The default transformations available have also been enhanced. As a result of these changes different molecules will be generated than with previous releases of the software.
When growing docked fragments, the conformation is randomly changed to attempt to remove CPK contacts with the protein and within the new molecule. In addition, target residues can be specified in the protein (in the same way as required interactions with pharmacophore searching under the Advanced tab of the FindSites node - see Identifying Atoms), and the generated molecules are assigned a merit score between 0 and 100 for each residue based on whether these interactions are achieved. The sum of the merit scores for each molecule-reside interaction is stored in a field named Merit. Saved conformations will not have contacts with the protein (because such molecules are rejected) and will have the geometry of highest scoring conformer sampled.
As with previous releases increasing the CURB value to allow more non-hydrogens atoms is important to generate molecules with a significantly higher molecular mass. Limiting substitution positions to those atoms belonging to a specific group can also be highly desirable.
The conformations of the generated molecules can be refined using the Docking node with the conformational generation option set to "None" to avoid redocking the molecules and the refinement set to "Side-chain and Ligand".
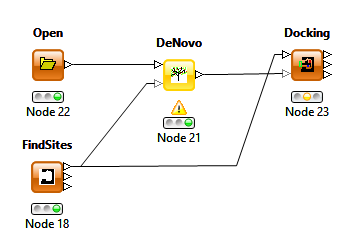
This release also includes the option to select the protein query generated by the Find Sites node under the THINK setup tab of the Docking and De Novo nodes removing the necessity of including a filter node.
The combinatorial chemistry enumeration has been extended for use in FBDD where the bound fragment can be used as the combinatorial core and R-groups are attached in specified position(s). Like the De Novo functionality, conformations are randomly sampled to avoid CPK contacts with the protein and to optimise interactions with desired residues. In addition to the protein, the implementation has the option for an orientation fragment which is typically part of the original docked fragment, to which the enumerated molecule is fitted. This approach allows greater flexibility in the choice of the core than retaining the original docking fragment but is slightly more complicated to configure.
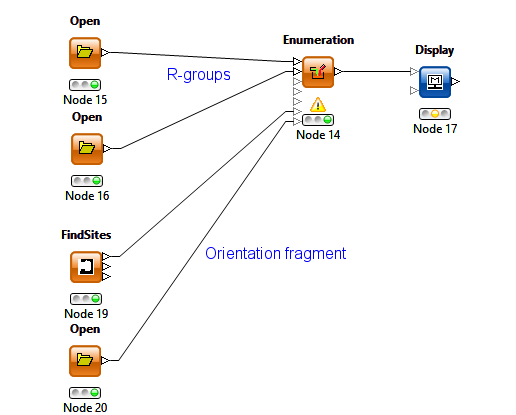
The KNIME enumeration node has been extended with the addition of 2 additional optional inputs for the protein query (created by the FindSites node) and the orientation fragment.
KNIME (pronounced naim) provides a means of visualising and configuring the steps for a molecular modelling study where each step has a pictorial representation as a node in a workflow. More information can be found here.
THINK 2.00a is compatible with KNIME 2.9.x and (hopefully) subsequent releases under Windows and Linux.
Note: It is necessary the define the environment variables THINK_EXEC and THINK_WORKING for the paths to the THINK software folder and a working folder. Different users may have different working folders. Without these definitions KNIME does not know the location of the THINK software or where to write temporary files for THINK.
There are 2 major changes:
Several smaller changes have been made including additional inputs on the enumeration node for FBDD (see above).
Enhancements to Pharmacophore and 3D Searching
Critical parts of the underlying code has been rewritten to allow for multi-threading of especially non-bonded interactions, general speed enhancements and accelerators for 2-centre pharmacophore searching (which were previously present only for 3 and 4 centre searching). Note: The parallel processing option is available only from KNIME and when it is used the hits are returned in a different order than that in the original file being searched. If no bond rotations are specified or the number of sample orientations per bond is set to 1 (recommended), then the orientation and torsions in the starting conformation are refined (this is important for refining the conformations of grown fragments using both the de novo and combinatorial chemistry methods). When using the KNIME docking node, it is recommended to set conformational generation to "none" to refine existing coordinates.
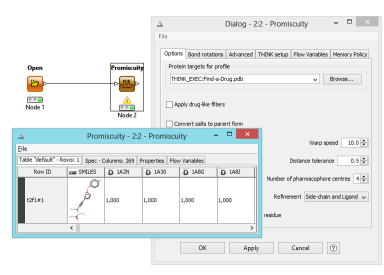
The promiscuity node performs a serious of pharmacophore searches for queries stored in a profile definition file in pdb format. The output consists of a table of scores for rows of molecules. Within the profile definition file, the number of pharmacophore centres, warp speed etc can be overriden as appropriate for each query.
THINK now handles text data in molecular data fields. This that when reading SD or SMILES files, text data fields are no longer discarded and will be displayed in worksheets and saved in files.
The maximum length of symbol translations has been increased allowing text strings up to 8192 characters to be stored.
Some PDB files contain duplicate atoms which are alternative positions for the same atom but have not been recorded correctly as such. These can cause connectivity and other problems and are now reported as errors on reading such PDB files.
X and Y flip and Z-rotate functionality has been added to 2D draw.
An obscure bug which caused some very hydrophobic molecules to generate an error when creating lipophilic centres has been fixed.
Reject and Learn default file names
The keyboard commands have been implemented and corresponding changes in KNIME nodes to allow users to specify the default rejection file (overriding reject.smi) and the default learn file (overriding default.lrn). This is inline with the earlier change which allowed the default transformation file used by the de novo derivative generation to be specified (overriding tranform.smi).
The common variable IERROR is now set to the most recent error number and the function $MESSAGE() will return the most recent error message text. This function can also take an argument to return the text associated with that error number.
When processing larger data sets, the time taken to create tables and load the data into KNIME is significant. Furthermore, some nodes use .csv files to transfer data back from THINK to KNIME which in previous releases always contained the 2D fingerprints for the molecules even if these were not required and had to be calculated specially. The new command line option NOKEYS omits these fingerprint keys from .csv files and this option is now used by most KNIME nodes to speed up the data import.
Some calculations such as those performed by the Promiscuity node can take a long time and in the event of a failure there is now an option to restart from the molecule being processed, avoiding repeating the entire calculation.
As a result of a customer request, the names of the plugins (.jar files) have been changed to conform the KNIME convention. They now start "com.treweren". Unfortunately, this means that existing workflows will need to be created as they are linked to the .jar file names not to the node names.
There are no software changes.
The User Guide has also been rewritten for this release.
KNIME (pronounced naim) provides a means of visualising and configuring the steps for a molecular modelling study where each step has a pictorial representation as a node in a workflow. More information can be found here.
THINK 1.42 allows optional inputs to be used in place of files for Search, Similarity, R-group and Docking nodes. In addition, it is now only necessary to connect those inputs of the Enumeration node which correspond to R-groups which are to be used in the enumeration.
THINK 1.42c is compatible with KNIME 2.2.0 and does not work with earlier releases.
Note: It is necessary the define the environment variables THINK_EXEC and THINK_WORKING for the paths to the THINK software folder and a working folder. Different users may have different working folders. Without these definitions KNIME does not know the location of the THINK software or where to write temporary files for THINK.
The 3D De Novo capabilities have been enhanced to allow docked fragments to grow into larger drug-like molecules. In this case, larger increases in the number of non-hydrogens atoms can be enabled by setting the CURB value. The transformation probability is progressively decreased for transformation which insert more than this number of non-hydrogen atoms to reach zero at twice the curb value.
The 3D display functionality has been enhanced by functionality which allows molecules to be orientated to view along a bond, a vector (line between 2 atoms) and onto a plane. The Orientate item is found on the right mouse button popup.
The ability to fit molecules together by matching pairs of atoms is now available from 3D display. The Superimpose item is found as a subitem to Orientate on the right mouse button popup. The functionality to manipulate a fragment independently of other fragments has also been included. Use the Global motion item on the right mouse button popup to switch between global motion and that of a selected fragment.
The select and highlight atoms has been extended to use modifier keys which are pressed at the same time as the mouse is clicked to allow:
| Select | Click | Modifier key |
| Atom | Atom | |
| Molecule | Atom | TAB |
| Fragment | Atom then Same atom | SHIFT |
| Path | First atom then Second atom | SHIFT |
| Bond/Chain | Bond | |
| Atoms in sphere | Click atom drag then Release | |
| Atoms in rectangle | Click drag then Release | |
| Invert selection | CTRL with above |
These capabilities can be used with the new Edit Group found on the right mouse button popup. Select the group number to which atoms are to be set then select the atoms (which will be highlighted). This functionality is useful when the 3D De Novo capabilities are used for controlling which parts of docked fragments grow into molecules.
Coordinates for most bridged rings can now be generated (2D and 3D) and adequately displayed.
There have been some improvements to 2D Edit including:
In most cases, it is no longer essential to use explicit hydrogens in De Novo transformations as these will be added with the appropriate group numbers automatically. When saving transformations using text commands a new option TRANSFORMS forces hydrogens to be included and converts atom type 0-9 for connections to A.
The functionality to create interaction centres for binding sites has been extended to include the creation of lipophilic centres.
There are no software changes.
KNIME (pronounced naim) provides a means of visualising and configuring the steps for a molecular modelling study where each step has a pictorial representation as a node in a workflow. More information can be found here.
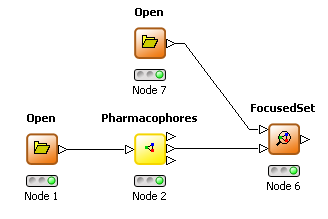
THINK 1.41 has two additional nodes to allow focused and diverse subsets of molecules to be selected based on the pharmacophores they exhibit compared to a reference set. The pharmacophore node has been enhanced by to allow the pharmacophore profile for a set of molecules to be an additional output. A focused selection has overlapping pharmacophores with this reference set whereas a diverse selection has non-overlapping pharmacophores. The minimum number of overlapping or non-overlapping pharmacophores may be configured.
| Workflow | Configuration | Workflow | Configuration | ||
 |
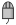 |
FocusedSet node |  |
|
DiverseSet node |
The De Novo node has also been enhanced to provide 3D De Novo capabilities.
Demonstration videos of the main new capabilities are available.
 | Focused subset |
| 3D De Novo |
THINK 1.4.1 is compatible with KNIME 1.3.3.
Note: It is necessary the define the environment variables THINK_EXEC and THINK_WORKING for the paths to the THINK software folder and a working folder. Different users may have different working folders. Without these definitions KNIME does not know the location of the THINK software or where to write temporary files for THINK.
The De Novo structure generation has been enhanced to modify/add 3D substituents to a molecule which has been previously docked into a protein site. Only transformations with one connection position are used whereas 2D De Novo derivative transformations may have multiple connection positions. This development requires the protein to be present in order to constrain the derivatives (and their conformations) in order to fit within the binding site. If derivatives of the known ligand from a PDB file are required, then this ligand can be extracted and saved (output 3 from the FindSites node) for subsequent use with the Docking node.
| Workflow | Configuration | |
 |
|
De Novo node |
| Commands | Dialogs | |
| SUGGEST MOLECULE=#1 MAXIMUM=100 TIME=1000 OUTPUT=more.sdf SITE=1FGI | 3D De Novo is not supported |
The 3D De Novo functionality may also be used with a pharmacophore volume constraint map. When using KNIME these can be generated by selecting the appropriate mode in the Pharmacophore node, using the Search node to orientate the molecule inside the constraint map and the corresponding mode in the De Novo node to use the map as a constraint.
| Workflow | Configuration | |
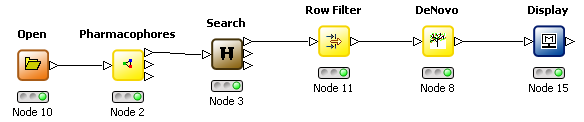 |
|
De Novo node |
Note: There have been changes to the transformation file (tranform.smi) which may result in different 2D derivatives being generated than earlier releases.
The map display visbility can now be toggled on the right mouse button pop-up menu without returning to the configuration dialog. This is especially useful when using a KNIME workflow.
The 3D display with a display panel has been enhanced by the addition of a scroll bar on the main graphics window. For a series of docked molecules/poses, this allows users to scroll between them without clicking on the Display Panel to swap visibility.
The sensitivity of the mouse movement for scaling and clipping has been reduced.
If the connectivity of a non-standard residue is omitted in a PDB file, THINK will now attempt to generate the connectivity (as well as the bond order).
CSV output files will no longer contain fields which have not been populated or previously referenced/defined.
The use and definition of added (ie implicit) hydrogens has been improved with the consequence that they are no longer included in MACCS SD 2D files which are subsequently written. (This was important for R-group searching and enumeration).
There are no software changes.
KNIME (pronounced naim) provides a means of visualising and configuring the steps for a molecular modelling study where each step has a pictorial representation as a node in a workflow. Using workflows for computer aided chemistry is both easier and more intuitive than traditional GUI and command scripts. Workflow technology is well established in the pharmaceutical industry for ChemInformatics applications usually processing 2D information, for instance to select subsets of commercial catalogues.
Demonstration videos of the main capabilities are available.
| 2D functionality |
| Structure-based virtual screening |
| Pharmacophore tools |
KNIME is also used by some other vendors of computational chemistry software and provides a platform for integrating functionality provided by vendors as well as developed in-house. KNIME also includes a wide-range of statistical tools. The automation of KNIME facilitates exploring more possibilities such as alternative binding sites and reporting the performance of different possibilities in concise tabular forms.
Note: It is necessary the define the environment variables THINK_EXEC and THINK_WORKING for the paths to the THINK software folder and a working folder. Different users may have different working folders. Without these definitions KNIME does not know the location of the THINK software or where to write temporary files for THINK.
In previous releases, water molecules could be included in the protein molecule associated with a site query. These molecules are ignored if they overlap with ligand atoms. Water molecules which are present in the binding site and interact with a ligand used to create the query are now automatically placed and saved in the protein molecule. This eliminates adding these waters by manually editing the PDB file.
When creating a site query using a docked ligand, it was possible that the interaction centre positions were not appropriate for reproducing a known interaction such as a hydrogen bond. If the distance between the relevant ligand atom and the nearest interaction centre is greater than the tolerance an additional interaction centre is now placed at the coordinates of the ligand atom. There have also been some small changes to the interaction centre positioning including increasing the separation of aromatic centres from the defining ring to 3.7Å and the number of centres and their positions for water molecules.
A command and dialog item has been added to allow distances to be listed. When used to list interaction distances between a ligand and a protein it is necessary to specify the molecules and desirable to specify a distance range to limit the size of the listing.
| Commands | Dialogs | |
| LIST INFO=DISTANCES SELECT=^2CHM,^#3,0,4 | List distances is not supported | |
| LIST INFO=DISTANCES SELECT=^2CHM,"^CHM-METAL",0,4 |
An option has been added to omit hydrogens from SD files. In previous releases 3D SD files always included hydrogens while 2D SD files do not.
| Commands | Dialogs | |
| SAVE FILE=molecule.sdf OPTION=NOHYDROGEN | Not supported from dialogs |
Functionality has been added to calculate bond orders retaining existing connections. This is used automatically when reading HET groups such as ligands from PDB files. The algorithm which is also used when computing connections has been improved to deduce aromaticity for planar rings.
| Commands | Dialogs | |
| MODIFY MOLECULE=#1 REBUILD=BOND-ORDERS | Not supported from dialogs |
The pharmacophore area and volume checks during a site searches eliminate hits which exhibit the pharmacophore in a small fraction of the molecule. This can now be disabled by setting the volume/area to zero giving more hits.
| Commands | Dialogs | |
| CUSTOM AREA=0. VOLUME=0. | Not supported from dialogs |
There have been a number of small changes which have improved the integration with KNIME and access to appropriate functionality:
There are no software changes.
Help text is now available for all dialogs as tooltip help (in a popup box) for each item. The help text for all items on a dialog is also available online and accessed by clicking Menus on the Help menu.
An extra tab has been added to the Preferences dialog for specifying the activity field, activity type and significance (to identify the most active and most inactive molecules). When the activity type is specified the significance configuration is changed. The functionality to subdivide molecules into "most active" and "most inactive" has been revised to allow absolute values of activity to be specified.
| Commands | Dialogs | |
| CUSTOM ACTIVITY=activity SIGNIFICANCE=4,8 ATTRIBUTES=HIGH | |
File > Preferences > Activity |
Previously, a subset of these activity configuration controls were duplicate in part on several different dialogs. In command mode, it is now necessary to use the CUSTOMISE command rather than additional keywords to various other commands.
Profile Comparisons
Logical operations between a profile being read and on already in memory may now be performed. Common pharmacophores between the profiles for two sets of molecules may be identified using AND and different pharmacophores using the NOT keyword. Other logic keywords include ADD, NEW, OR and EOR.
| Commands | Dialogs |
| READ FILE=profile1.pro | Profile logical operations not supported |
| READ FILE=profile2.pro LOGIC=AND |
Saving Pharmacophore Coordinates
The coordinates for the pharmacophores in a profile can now be stored in a file with the extension .phc. This change allows such files to be created without resorting to command scripts. It is usually adviseable to limit the number of pharmacophores written and/or specify a threshold on the pharmacophore population. The profile is ordered so that pharmacophores with the highest population are written first.
| Commands | Dialogs | |
| CUSTOMISE PHARMACOPHORE=100 EMPTY=10 | |
File > Preferences > Pharmacophores |
| SAVE FILE=pharm_coords.phc molecule=* | |
File > Save As > Type: Pharmacophore coordinates |
Please note: In general, it is advisable to specify the molecule=* option in command mode as otherwise an existing profile will be used to avoid unnecessary recalculation.
Activity Profiles
If the name of the field containing the activity and an activity threshold (with associated attributes) is defined, then the profile calculated is that for the active molecules is less that for the inactive molecules. In THINK 1.32g the profiles for the active and inactive molecules are normalised before subtraction.
| Commands | Dialogs |
| CUSTOM ACTIVITY=pactivity ATTRIBUTE=ABSOLUTE SIGNIFICANCE=6 | Not supported via preferences |
| SAVE FILE=profile1.pro molecule=* |
Pharmacophore Profiling Performance
The performance of pharmacophore profiling has been improved by applying pharmacophore volume and accessibility tests to the pharmacophores. This reduces the number of small and irrelevant pharmacophores in the profile and significantly improves the performance by typically more than 10-fold.
If one or more molecules do not exhibit any pharmacophores, then a trace option can be used to report the reasons to the console. (The same trace mode is supported for 3D searches).
| Commands | Dialogs |
| LET #ITRACE=1025 | Not supported |
The steps necessary to create a Ligand Query Volume map from a set of active molecules and use this in 3D pharmacophore searches with volume constraints have been brought together in a single dialog. Previously it was necessary to perform the following tasks using different dialogs or hand written command scripts:
The new dialog has the essential configuration options and 3 buttons to perform the tasks in sequence. The first step is invariably to specifiy the file containing the active molecules. Additional information about the calculations is written to a file with the same filename as the active molecules and the extension type .lis. In most cases it is advisable to limit the number of pharmacophores saved to around 100 and ensure that the validation percentage allows one of the active molecules not to be found. It is not unusual to increase the tolerance to 1.
| Commands | Dialogs | |
| No command equivalent | |
Calculate > Ligand Query |
If activity data is present in the molecule file, the activity field name, type and significant configured, then a difference pharmacophore profile will be calculated which can reduce the number of pharmacophores and maps which are created. When this file is used in the search step, then additional output reports how successful each volume map is at eliminating inactive molecules and future searches can be restricted to the most effective map constraint.
Known limitation: The file containing the active molecules must be in the current working folder. If this is not the case then the second step fails with a file not found message.
A number of improvements have been made to the scoring function which are referred to as THINKScore 1.0 (abbreviated to TS1.0) which is used by default in preference to ChemScore 2.1 (abbreviated to CS2.1) which remains available. The most significant difference is the use of a Coulombic charge term for hydrogen bonding in preference to the ChemScore parameterised term.
| Commands | Dialogs |
| CUSTOM SCORE=CS2.1 | Only changed using commands |
The constants used in the scoring function are in common variables (reported by LIST CUSTOMISE) and in some circumstances it may be appropriate to change these values using command scripts.
A new option has been added to adjust the side-chain torsion angles of residues in the receptor site. In practice, this is a better alternative than using soft VdW interactions that allow false positives to fit. The necessary changes to the VdW terms of the scoring function and a switch to using a Steepest Descents (rather than Conjugate Gradient) minimiser is performed automatically when side-chain motion is enabled.
| Commands | Dialogs | |
| SEARCH MODE=SITE FILE=1bqo-q1-tested.smi QUERY=1BQO-Q1 OPTION=SIDE-CHAIN | |
Search > 3D > Side-chains |
Multiple Volume Map Search Constraints
THINK 1.32 has a subtle but important improvement when using volume map search constraints. Previously, it was only possible to use one volume map for each pharmacophore during the search although the code which generated these maps produced several alternative volume map constraint for each pharmacophore. If a single map is specified using the SITE keyword, then only that map is used in the search otherwise all ligand maps are used as search constraints. The consequence of this enhancement is that the time for performing searches with map constraints is significantly reduced because the need to perform multiple searches with the same pharmacophore is eliminated.
Molecules which are very flexible are often filtered out by "drug-likeness" criteria. However, on occasions it may preferred to use a sampling method for the more flexible molecules. THINK 1.32 can be configured to automatically swap from systematic conformation generation mode to sampling such molecules based on the number of rotating bonds or an estimate the number conformations which will be processed (ie the flexibility derived from the product of the number of sample points about each bond). This enhancement means that slightly more flexible molecules can be included pharmacophore profiles without excessive time penalties.
| Commands | Dialogs | |
| CUSTOMISE TWIST=8 GENERATE=6561 SAMPLE=1000 | |
File > Preferences > Conformers |
A command and dialog item has been added to allow partial atomic charges to be listed (these are used by the THINK scoring function). The partial charges are calculated by the Skorcyzk method as this gives the best hydrogen bonding energies.
| Commands | Dialogs | |
| LIST INFO=CHARGES MOLECULE=#1 | |
View > Atom Data > Data: Charges; List |
The ROTATE command has been extended to apply a global rotation to generate the best plane projection for a map. This orientates the map so that its longest inertial axis is along the X-axis (horizontal) and shortest axis is along the Z-axis (out of the screen). Similar functionality exists for a molecule or list of atoms.
| Commands | Dialogs |
| ROTATE TOMAP=mapname | Not available from dialogs |
When executing a command script, an extended progress dialog is now displayed which includes the file name of the script and the fraction of the file processed. The default description can be overridden using the symbol DESCRIPTION and the default fraction complete can be overridden using the symbol PROGRESS. The default behaviour may be restored with the value FILE (or undefined) and the progress dialog omitted if the DESCRIPTION value is set to OMIT.
The THINK interpreter gave priority to add before subtract in arithmetic expression which does not give the intuitive results. Priority is now given to subtract!
An obscure precision problem affected some Boltzman population pharmacophore profiles, when the populations and the denominator (or partition function) was not computed with sufficient precision. In THINK 1.32 relative energies are used for calculating the exponent term and the base energy value is reset during the calculation when appropriate.
In pharmacophore profiles, the maximum population of a bin is now 100% regardless of redundant populations (when one conformation exhibits the a pharmacophore more than once).
There are no software changes.
The graphics capabilities have been enhanced by the ability to display and manipulate 3D VdW volume and electrostatic potential maps. Users may specify the level and colours of the contours and delete the maps when they are no longer required.
| Commands | Dialogs | Output | |
| CALCULATE MAP=dopamine_VDW TYPE=VDW MOLECULE=dopamine | |
Calculate > Maps > Calculate | |
| CUSTOM VDW=1.0,Magenta,0.99,Blue | | File > Preferences Contours | |
| DISPLAY MODE=3D STYLE=STICK MOLECULE=* MAP=dopamine_VDW | |
View > Molecule 3D Stick; Show Map:dopamine_VDW; Display | 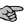 |
| LIST INFO=MAPS SELECT=T11_* | |
View > Atom Data > Data: Maps; List | |
| DELETE MAP=T11_* | |
Edit > Delete > Maps; Delete |
Maps may also be saved to disk with the file type .map when they are written as a text file which can be read in on another occasion. It is important to appreciate that when a global viewing transformation is applied, the orientation of any map subsequently saved will be changed. This means that if molecules and maps are saved at different times with intervening transformations, they will not be aligned correctly when read back in!
| Commands | Dialogs | |
| SAVE FILE=test2.map MAP=T11_* | |
File > Save As > Save As Type: Maps; File name: test2.map; Save |
The current implementation does not include the ability to perform logical operations between maps such as Union (OR), Difference (XOR) etc
Active Volume Search Constraints
This major enhancement allows Site Searches to be performed without a model or crystal structure of the active site. In overview, a volume constraint which represents the known minimum volume of the binding cavity is automatically constructed from a set of known active molecules. The implementation uses a combination of the pharmacophore profiling capabilities and the 3D volume maps.
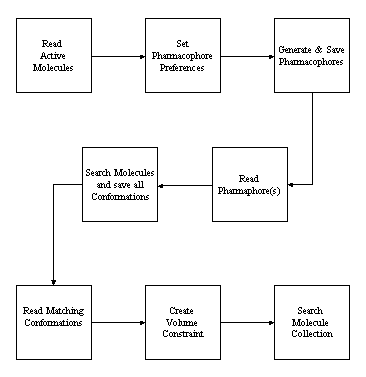
When the pharmacophore profile is exported with the file type .phc as a set of coordinates for each pharmacophore, it is sorted by population so that the most common pharmacophores across the set of active molecules are written first. When using dialogs, the pharmacophores are calculated and saved for all molecules using the current conformational generation and pharmacophore configurations. Occasionally, the population exceeds 100% which implies that at least one molecule exhibits that pharmacophore more than once in the same conformation. The pharmacophores are sorted in descending population count and it is often useful to truncate the number saved either by the ignoring counts below a threshold value (the empty value) or by processing the 'n' most common pharmacophores.
| Commands | Dialogs | |
| CUSTOM COUNT=POPULATION EMPTY=10 PHARMACOPHORES=100 | |
File > Preferences Pharmacophores Profile:Boltzman Empty=10; Use best=100 |
| SAVE FILE=actives.phc | |
File > Save As > Save As Type: Pharmacophore coordinates; File name: actives.phc; Save |
The pharmacophores can be used immediately in a Site Search but in the absence of a volume constraint tend to give too many false positives. It is often necessary to increase the default tolerance (of 0.5) in order to validate that the set of molecules used to generate the pharmacophore, will be found in a Site Search using that pharmacophore. This is an inevitable consequence of the granularity of the bin model used to represent distances in the pharmacophore profile. There are usually a number of pharmacophores which occur sufficiently frequently to justify their use as search queries.
When a pharmacophore is used in a 3D Search, a set of conformations is generated which exhibit that query. These conformations are used to calculate a volume search constraint for that pharmacophore. The software will automatically select subsets of these conformations (one for each molecule) which exhibit the lowest union volumes and create a volume map for each subset and (by default) retain the best 10 alternative maps distinguished by numeric suffixes. If no molecules are specified, all molecules with conformation numbers are processed. Such ligand volume maps can be used as active volume search constraints analogously to the protein residues which constitute the active site.
| Commands | Dialogs | |
| SEARCH MODE=3D QUERY=ADDX-115266 FILE=actives.smi OUTPUT=addx-115266.sdf OPTION=ALL-CONFORMERS | |
Search > File: actives.smi; Best Conformation:Off; Search |
| CALCULATE MAP=ADDX-115266-LIGAND TYPE=LIGAND | |
Calculate > Maps > Ligand; Map name: ADDX-115266-LIGAND; Calculate |
| SEARCH MODE=SITE QUERY=ADDX-115266 FILE=collection.smi OUTPUT=hits.sdf | |
Search > File: collection.smi; Site:On; Search |
In a site search, if the name of the constraint map is omitted, the first active volume map present will be used although the first protein site will be used in preference if present.
The 3D display manipulation has been enhanced to allow the clipping slab thickness to be adjusted using Y-motion of the mouse while holding down the TAB key.
There are no software changes.
This file contains the release notes for THINK v1.30 and THINK SERVER v1.30.
Some molecular mechanics capabilities have been added. These include a Simplex geometry optimisation and the use of the molecular mechanics energy in pharmacophore profiling (see below). In general it is NOT recommended to optimise the geometry of the starting conformation of a molecule in a conformational search as this inevitably gives a bias to the conformations which are accepted. However, the geometries of strained rings are usually improved by a molecular mechanics geometry optimisation and then fixed during a conformer generation.
| Command: | MODIFY REBUILD=OPTIMISE MOLECULE=name |
| Dialog: | Not available from dialogs |
The molecular mechanics energy calculation use simple (Chem-X like) quadratic terms for bond length and angle cosine terms. The form and barrier constant of the torsion term depends on whether the bond is conjugated or not. A Lennard-Jones (12-6) term is used for non-bonding interactions. The constants were included in the parameters files of previous versions. The current implementation has no option to include a Coulombic energy term arising from partial charges.
| Bond length | kl(l-lo)2 | Where l is the bond length, kl and lo are constants for each pair of atom types |
| Bond angle | kc(c-co)2 | Where c is the cosine of the angle, kc and co are constants for each atom type |
| Conjugated torsion | Vc(1-cos2f) | Where Vc is the conjugated torsion constant and f is the torsion angle |
| Non-conjugated torsion | Vnc(1 + cos3f) | Where Vnc is the non-conjugated torsion constant and f is the torsion angle |
| Non-bonding interactions | e1e2(1/r12 - 2/r6) | Where e1 and e2 are constants for the atom types and r is the distance between them |
At present, the molecular mechanics energy cannot be reported for a single conformation of a molecule or included in the property table.
The PDB format does not explicitly support the division of its contents into molecules. There are usually several indicators which can be used to identify molecules but the process is prone to error because certain types of records (eg HETNAM and HETSYN) are used for both non-standard residues and non-protein molecules. A strict application of the molecule concept would also create a separate molecule every occurrence of water and make somewhat arbitray decisions (based on connectivity) about whether a residue chain should be a different molecule.
For greater convenience THINK 1.30 attemps to create the following molecules from a PDB file:
In addition, changes have been made to reading site and secondary structure records.
| Command: | OPEN FILE=filename CHAIN=SEPARATE |
| Dialog: | Not available from dialogs |
Under some circumstances it is still necessary to edit a PDB file manually and insert NAME records to indicate the start of each molecule. For instance, when certain water molecules which are present in the active site are essential for binding and need to be in the protein molecule or when residue chains need to be grouped into subsets and stored as molecules.
The way in which the site definitions are stored has been changed and the site identifier is now included in the residue part of the atom specification following the sequence number and separated by a full stop eg _(123A.ST1). Any secondary structure assignment is also included after the chain identify and separated from it by a full stop. Some new keyword values have been added to the INFO parameter of the LIST verb.
| SITES | To list the protein binding sites |
| RESIDUES | To list the residues specified using the SELECT keyword |
| ATOMS | To list the atoms specified using the SELECT keyword |
| Command: | LIST INFO=SITES |
| Dialog: | View > Atom Data > Data : Sites, List |
Pharmacophore Population Profiles
The original implementation of pharmacophore profiling counts pharmacophore occurrences without consideration of the population of the conformers in which the pharmacophore occurs. This has the potential to cause misleading comparisons of pharmacophore profiles. In addition, it is not apparent from the pharmacophore count the proportion of molecules that exhibit a pharmacophore.
Two new options have been added to the CUSTOMISE COUNT keyword in 1.30:
| Command: | CUSTOMISE COUNT=POPULATION |
| Dialog: | File > Preferences > Pharmacophores > Pharmacophore Profile: Boltzman Population |
Note: If a pharmacophore occurs more than once in a conformation, this can lead to the profile value exceeding 100%.
Additional functionality has been added to generate and write out the pharmacophore geometries in SD file format.
| Command: | SAVE FILE=filename OPTION=COORDINATES |
| Dialog: | Not available from dialogs |
The functionality to select a subset of diverse molecules has been extended to allow pharmacophores to be used. The minimum number of new pharmacophores is expressed in percentage points when using POPULATION or FRACTION count methods.
| Command: | SELECT FILE=filename OUTPUT=filename PHARMACOPHORES=n |
| Dialog: | Calculate > Selection > Pharmacophores |
After some hardware failures or shutdowns without stopping THINK, the progress information saved in the checkpoint or restart file was not consistent with the contents of the results file. In 1.30, the contents of the results file takes priority over the restart file, thus avoiding omitting hits in the results file.
The functionality of the Display Panel has been extended for proteins to include options to extend the current selection from typically one residue to the chain or site (if any) containing the original selection.
A minor correction has been made to the Lipophilic and Bad Contact terms in the docking scoring function. This can sometimes change the number of hits given by a search.
In the Windows version, the 2D functional group keys were sometimes written out as a string of question marks instead of hex codes. This was due to a compiler error and has now been worked around.
Progress reports have been added to the clustering code.
The queue locking implementation has been enhanced to support multiple copies of the queue manager running at the same time. This allows a copy to be running to assign jobs at the same time as another copy is used to load more jobs etc.